
Molecular Insight into the Self-Assembly Process of Cellulose Iβ Microfibril



Abstract
:1. Introduction
2. Result and Discussion
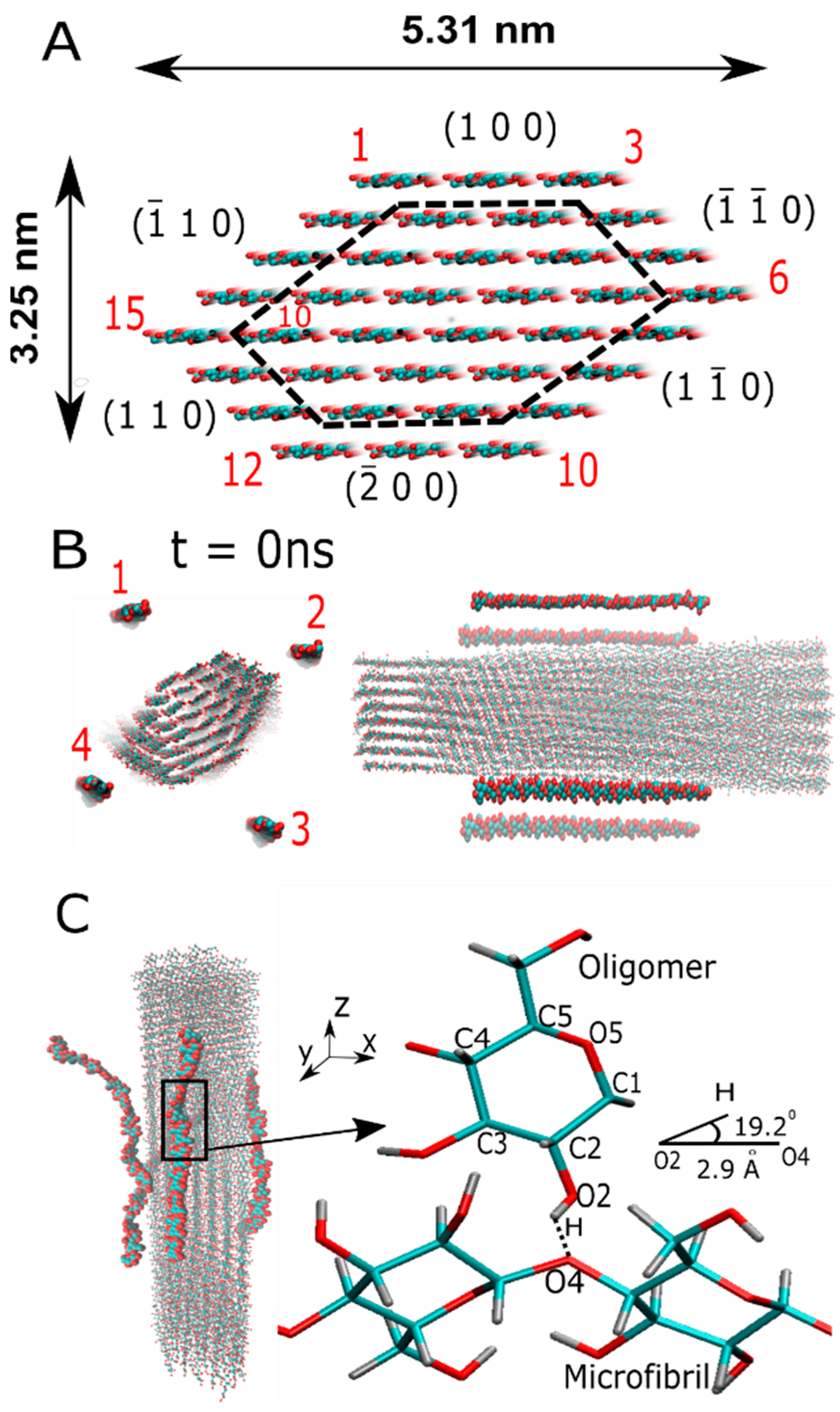
2.1. Oligomer–Microfibril Interaction: Relevance of the Chain Stiffness during Binding
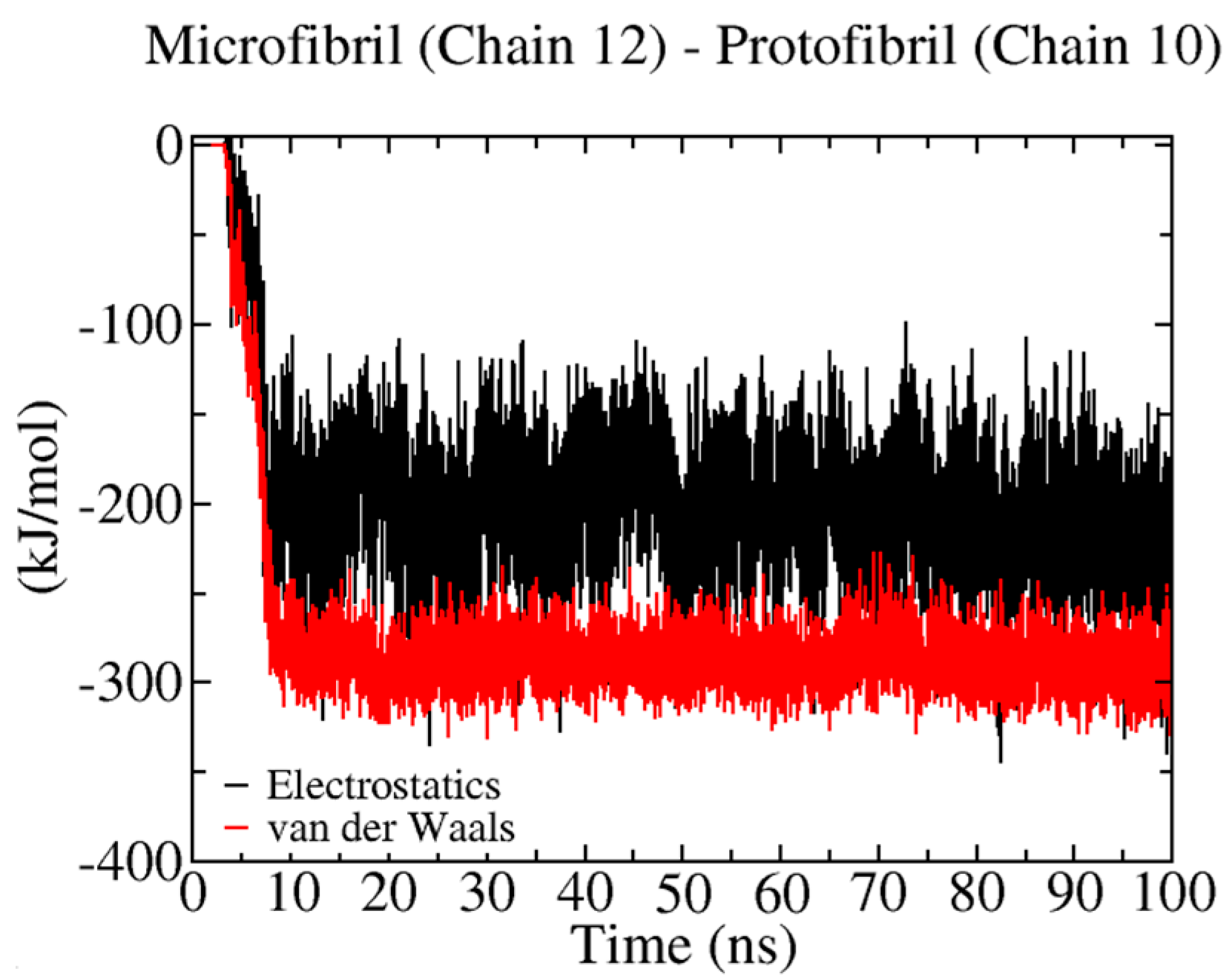
2.2. Energetic Characterization of the Binding Process: Electrostatic and vdW Energies
2.3. Hydrogen Bond Analysis
3. Materials and Method
3.1. Initial Structures
3.2. All-Atom MD Simulation
3.3. Energetic Analysis of the Aggregation Pathway
3.4. Fluctuation Analysis of the Bound Complex by Root Mean Square Fluctuation
3.5. Hydrogen Bond Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Purushotham, P.; Ho, R.; Zimmer, J. Architecture of a Catalytically Active Homotrimeric Plant Cellulose Synthase Complex. Science 2020, 369, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Jacek, P.; Dourado, F.; Gama, M.; Bielecki, S. Molecular Aspects of Bacterial Nanocellulose Biosynthesis. Microb. Biotechnol. 2019, 12, 633–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Turner, S. Plant Cellulose Synthesis: CESA Proteins Crossing Kingdoms. Phytochemistry 2015, 112, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal Structure and Hydrogen-Bonding System in Cellulose Ibeta from Synchrotron X-Ray and Neutron Fiber Diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Sugiyama, J.; Chanzy, H.; Langan, P. Crystal Structure and Hydrogen Bonding System in Cellulose I(alpha) from Synchrotron X-Ray and Neutron Fiber Diffraction. J. Am. Chem. Soc. 2003, 125, 14300–14306. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.S.; Chu, J.-W. On the Molecular Origins of Biomass Recalcitrance: The Interaction Network and Solvation Structures of Cellulose Microfibrils. J. Phys. Chem. B 2010, 114, 13333–13341. [Google Scholar] [CrossRef] [PubMed]
- Mostofian, B.; Smith, J.C.; Cheng, X. The Solvation Structures of Cellulose Microfibrils in Ionic Liquids. Interdiscip. Sci. 2011, 3, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Beckham, G.T.; Matthews, J.F.; Peters, B.; Bomble, Y.J.; Himmel, M.E.; Crowley, M.F. Molecular-Level Origins of Biomass Recalcitrance: Decrystallization Free Energies for Four Common Cellulose Polymorphs. J. Phys. Chem. B 2011, 115, 4118–4127. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Makshakova, O. Multifaceted Computational Modeling in Glycoscience. Chem. Rev. 2022. [Google Scholar] [CrossRef]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Coarse-Grained Model of the Native Cellulose Iα and the Transformation Pathways to the Iβ Allomorph. Cellulose 2016, 23, 1573–1591. [Google Scholar] [CrossRef] [Green Version]
- Moreira, R.A.; Weber, S.A.L.; Poma, A.B. Martini 3 Model of Cellulose Microfibrils: On the Route to Capture Large Conformational Changes of Polysaccharides. Molecules 2022, 27, 976. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Himmel, M.E.; Crowley, M.F. The Molecular Origins of Twist in Cellulose I-Beta. Carbohydr. Polym. 2015, 125, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Bellesia, G.; Chundawat, S.P.S.; Langan, P.; Redondo, A.; Dale, B.E.; Gnanakaran, S. Coarse-Grained Model for the Interconversion between Native and Liquid Ammonia-Treated Crystalline Cellulose. J. Phys. Chem. B 2012, 116, 8031–8037. [Google Scholar] [CrossRef]
- Abraham, E.; Deepa, B.; Pothan, L.A.; Jacob, M.; Thomas, S.; Cvelbar, U.; Anandjiwala, R. Extraction of Nanocellulose Fibrils from Lignocellulosic Fibres: A Novel Approach. Carbohydr. Polym. 2011, 86, 1468–1475. [Google Scholar] [CrossRef]
- Amin, F.R.; Khalid, H.; Zhang, H.; Rahman, S.U.; Zhang, R.; Liu, G.; Chen, C. Pretreatment Methods of Lignocellulosic Biomass for Anaerobic Digestion. AMB Express 2017, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- van der Lubbe, S.C.C.; Fonseca Guerra, C. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem. Asian J. 2019, 14, 2760–2769. [Google Scholar]
- Dudefoi, W.; Dhuiège, B.; Capron, I.; Sèbe, G. Controlled Hydrophobic Modification of Cellulose Nanocrystals for Tunable Pickering Emulsions. Carbohydr. Polym. Technol. Appl. 2022, 3, 100210. [Google Scholar] [CrossRef]
- Nishiyama, Y. Molecular Interactions in Nanocellulose Assembly. Philos. Trans. R. Soc. A 2018, 376, 20170047. [Google Scholar] [CrossRef]
- Moraïs, S.; Stern, J.; Kahn, A.; Galanopoulou, A.P.; Yoav, S.; Shamshoum, M.; Smith, M.A.; Hatzinikolaou, D.G.; Arnold, F.H.; Bayer, E.A. Enhancement of Cellulosome-Mediated Deconstruction of Cellulose by Improving Enzyme Thermostability. Biotechnol. Biofuels 2016, 9, 164. [Google Scholar] [CrossRef] [Green Version]
- Nidetzky, B.; Zhong, C. Phosphorylase-Catalyzed Bottom-up Synthesis of Short-Chain Soluble Cello-Oligosaccharides and Property-Tunable Cellulosic Materials. Biotechnol. Adv. 2021, 51, 107633. [Google Scholar] [CrossRef]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Elastic moduli of biological fibers in a coarse-grained model: Crystalline cellulose and β-amyloids. Phys. Chem. Chem. Phys. 2017, 19, 28195–28206. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.M.; Himmel, M.E.; Crowley, M.F.; Beckham, G.T. Decrystallization of Oligosaccharides from the Cellulose Iβ Surface with Molecular Simulation. J. Phys. Chem. Lett. 2011, 2, 1546–1550. [Google Scholar] [CrossRef]
- Bergenstråhle, M.; Wohlert, J.; Himmel, M.E.; Brady, J.W. Simulation Studies of the Insolubility of Cellulose. Carbohydr. Res. 2010, 345, 2060–2066. [Google Scholar] [CrossRef]
- Penttilä, P.A.; Paajanen, A.; Ketoja, J.A. Combining Scattering Analysis and Atomistic Simulation of Wood-Water Interactions. Carbohydr. Polym. 2021, 251, 117064. [Google Scholar] [CrossRef]
- French, A.D. Idealized Powder Diffraction Patterns for Cellulose Polymorphs. Cellulose 2014, 21, 885–896. [Google Scholar] [CrossRef]
- Ding, S.Y.; Zhao, S.; Zeng, Y. Size, shape, and arrangement of native cellulose fibrils in maize cell walls. Cellulose 2014, 21, 863–871. [Google Scholar] [CrossRef]
- Zhang, T.; Zheng, Y.; Cosgrove, D.J. Spatial organization of cellulose microfibrils and matrix polysaccharides in primary plant cell walls as imaged by multichannel atomic force microscopy. Plant J. 2016, 85, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Purushotham, P.; Fang, C.; Maranas, C.; Díaz-Moreno, S.M.; Bulone, V.; Zimmer, J.; Kumar, M.; Nixon, B.T. Synthesis and self-assembly of cellulose microfibrils from reconstituted cellulose synthase. Plant Physiol. 2017, 175, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Gomes, T.C.F.; Skaf, M.S. Cellulose-Builder: A Toolkit for Building Crystalline Structures of Cellulose. J. Comput. Chem. 2012, 33, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W., II; Mackerell, A.D., Jr. CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate-Protein Modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, E.; Abraham, M.; Hess, B.; Van der Spoel, D. Gromacs 2020 Manual. Available online: https://zenodo.org/record/3562512#.YuZU_hxBxPY (accessed on 14 July 2022).
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, M.I.; Poma, A.B.; Okazaki, K.I. Optimizing Gō-MARTINI coarse-grained model for F-BAR protein on lipid membrane. Front. Mol. Biosci. 2001, 8, 619381. [Google Scholar] [CrossRef]
- Liu, Z.; Moreira, R.A.; Dujmović, A.; Liu, H.; Yang, B.; Poma, A.B.; Nash, M.A. Mapping mechanostable pulling geometries of a therapeutic anticalin/CTLA-4 protein complex. Nano Lett. 2021, 22, 179–187. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligomer | Chain in Microfibril | HB Type (Donor-Acceptor) |
|---|---|---|
| Trajectory 1 | ||
| 1 | 8 | O6H⋯O2 [F5–I3] O2H⋯O6 [I1–F3] O6H⋯O2 [I2–F4] O2H⋯O6 [I5–F7] |
| Trajectory 2 | ||
| 4 | 18 | O3-H⋯O6 [F1–I6] O6-H⋯O3[F4–I3] O2-H⋯O6[F5–I2] O6-H⋯O3[F6–I1] O2-H⋯O6 [I5–F2] O2-H⋯C6 [I5–F2] O6-H⋯O3 [I2–F5] |
| Oligomer | Chain in Microfibril | HB Type (Donor-Acceptor) | |
|---|---|---|---|
| 4 | 15 | O2H⋯O6 [F15–I1] O2H⋯O2 [F17–I2] O2H⋯O4 [F23–I9] O2H⋯O1 [F27–I12] O3H⋯O3 [I2–F17] O2H⋯O3 [I4–F19] | C5H⋯O4 [I11–F26] C4H⋯O4 [I1–F16] |
| 4 | 16 | O2H⋯O3 [F15–I1] O2H⋯O6 [F19–I5] O2H⋯O6 [F25–I11] O2H⋯O6 [I1–F16] O2H⋯O6 [I4–F18] O3H⋯O6 [I6–F20] O2H⋯O6 [I10–F24] O2H⋯O6 [I12–F26] | |
| Oligomer | Chain in Microfibril | Type (Donor-Acceptor) | |
|---|---|---|---|
| Trajectory 1 | |||
| 1 | 1 | O2H⋯O3 [F13–I2] O6H⋯O2 [F14–I2] O6H⋯O2 [F22–I10] O2H⋯O6 [F25–I14] O6H⋯O2 [F32–I20] O4H⋯O6 [I1–F12] O3H⋯O6 [I10–F21] O2H⋯O4 [I12–F24] O6H⋯O6 [I16–F28] | C2H⋯O3 [I5–F17] C1H⋯O6 [I6–F18] C5H⋯O6 [I6–F18] C2H⋯O4 [F19–I8] C2H⋯O4 [I14–F26] |
| 2 | 13 | O3H⋯O3 [I8–F19] O6H⋯O2 [I14–F25] O6H⋯O3 [I16–F27] O6H⋯O3 [I20–F31] O3H⋯O6 [F17–I6] O2H⋯O2 [F21–I10] O2H⋯O2 [F23–I12] O6H⋯O6 [F24–I13] O2H⋯O3 [F25–I15] O6H⋯O4 [F28–I18] | |
| Trajectory 2 | |||
| 1 | 3 | O2H⋯O3 [I2–F17] O6H⋯O4 [I3–F18] O2H⋯O3 [I4–F19] O6H⋯O4 [I5–F20] O2H⋯O3 [I6–F21] O2H⋯O3 [I8–F23] O2H⋯O3 [I10–F25] O6H⋯O4 [I11–F26] O2H⋯O3 [I12–F27] O6H⋯O4 [I13–F28] O2H⋯O3 [I14–F29] O2H⋯O3 [I16–F31] O6H⋯O4 [I19–F34] O6H⋯O3 [F18–I4] O6H⋯O3 [F20–I6] O6H⋯O3 [F22–I8] O6H⋯O3 [F24–I10] O6H⋯O3 [F28–I14] O3H⋯O2 [F33–I18] | C4H⋯O2 [F35–I20] |
| 2 | 13 | O6H⋯O3 [I9–F19] O6H⋯O2 [I18–F27] O6H⋯O2 [F18–I8] O6H⋯O4 [F20–I11] | |
| Chain in Protofibril | Chain in Microfibril | HB Type (Donor-Acceptor) | |
|---|---|---|---|
| 7 | 12 | O6H⋯O4 [P2–F13] O6H⋯O4 [P4–F15] O6H⋯O4 [P8–F19] O6H⋯O4 [P10–F21] O6H⋯O4 [P12–F23] O6H⋯O4 [P14–F25] O6H⋯O4 [P16–F27] O6H⋯O4 [P18–F29] O6H⋯O2[F28–P17] | C4H⋯O2 [F14–P3] C1H⋯O2 [F27–P17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thu, T.T.M.; Moreira, R.A.; Weber, S.A.L.; Poma, A.B. Molecular Insight into the Self-Assembly Process of Cellulose Iβ Microfibril. Int. J. Mol. Sci. 2022, 23, 8505. https://doi.org/10.3390/ijms23158505
Thu TTM, Moreira RA, Weber SAL, Poma AB. Molecular Insight into the Self-Assembly Process of Cellulose Iβ Microfibril. International Journal of Molecular Sciences. 2022; 23(15):8505. https://doi.org/10.3390/ijms23158505
Chicago/Turabian StyleThu, Tran Thi Minh, Rodrigo A. Moreira, Stefan A. L. Weber, and Adolfo B. Poma. 2022. "Molecular Insight into the Self-Assembly Process of Cellulose Iβ Microfibril" International Journal of Molecular Sciences 23, no. 15: 8505. https://doi.org/10.3390/ijms23158505
APA StyleThu, T. T. M., Moreira, R. A., Weber, S. A. L., & Poma, A. B. (2022). Molecular Insight into the Self-Assembly Process of Cellulose Iβ Microfibril. International Journal of Molecular Sciences, 23(15), 8505. https://doi.org/10.3390/ijms23158505