
Hyperglycemia in Pregnancy-Associated Oxidative Stress Augments Altered Placental Glucose Transporter 1 Trafficking via AMPKα/p38MAPK Signaling Cascade

Abstract
:1. Introduction
2. Results
2.1. Participant Characteristics
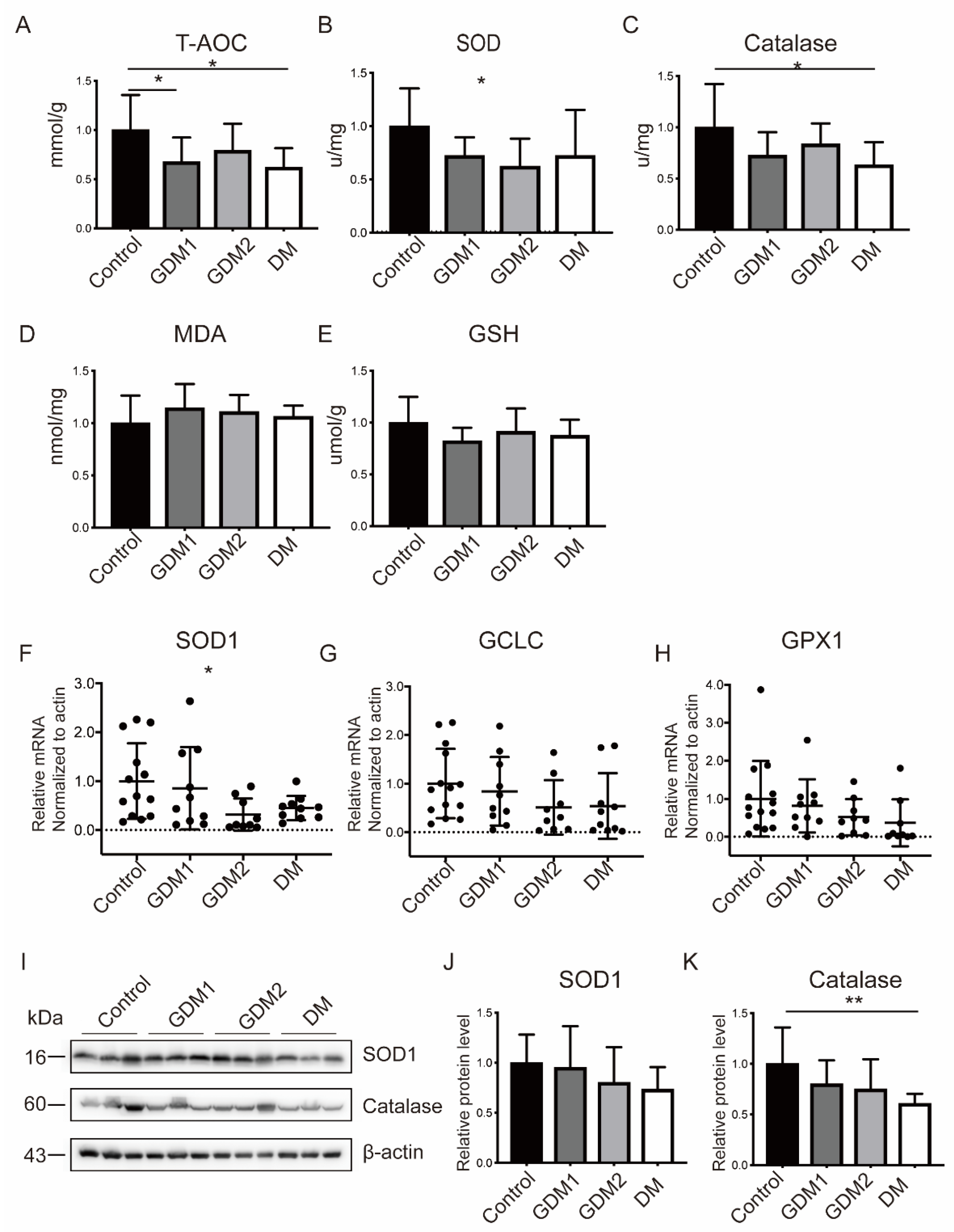
2.2. The Antioxidant Capacity Was Compromised in GDM and PGDM Pregnancies
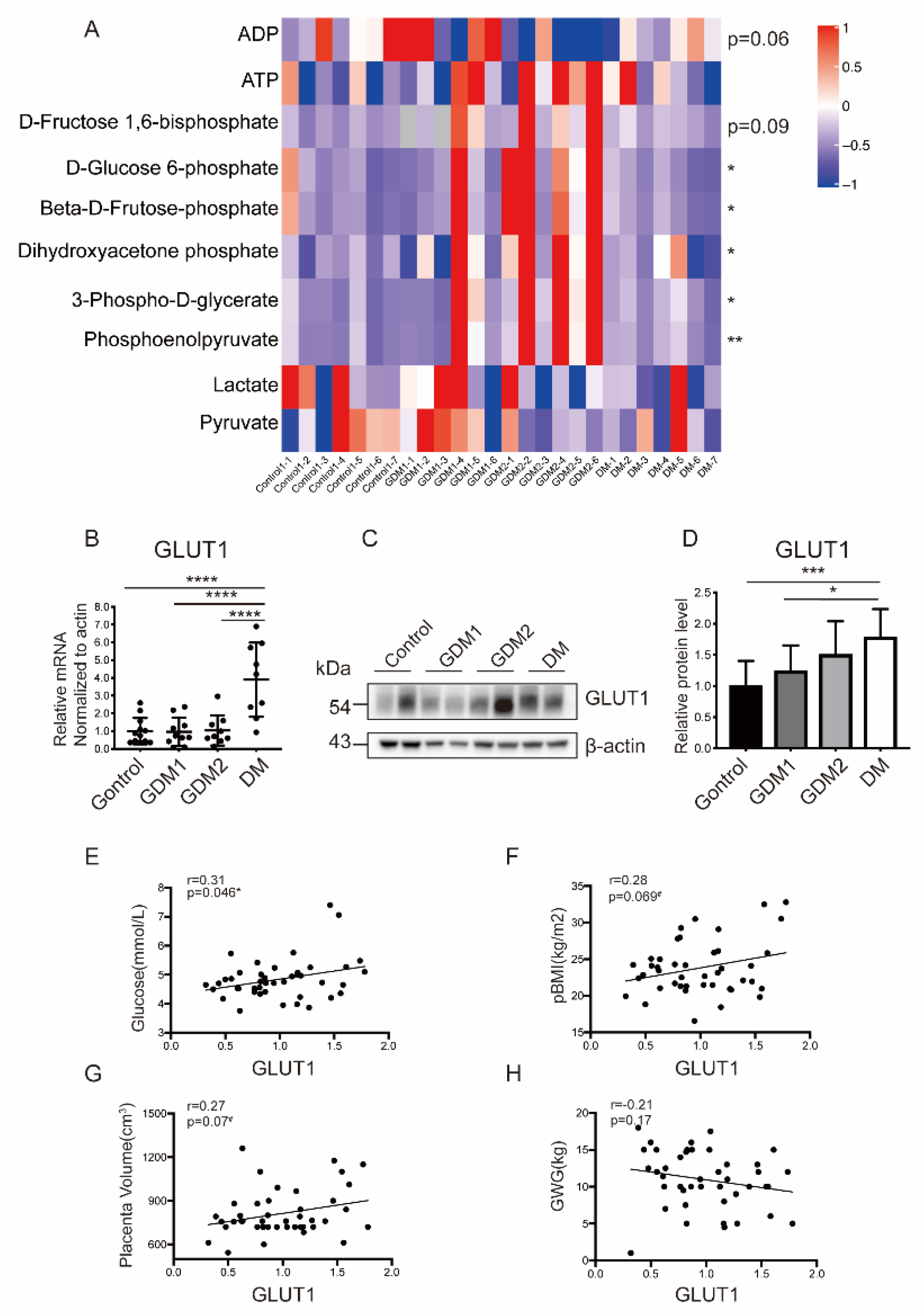
2.3. Glucose Metabolism Was Disrupted in Placentas of Women with Hyperglycemia
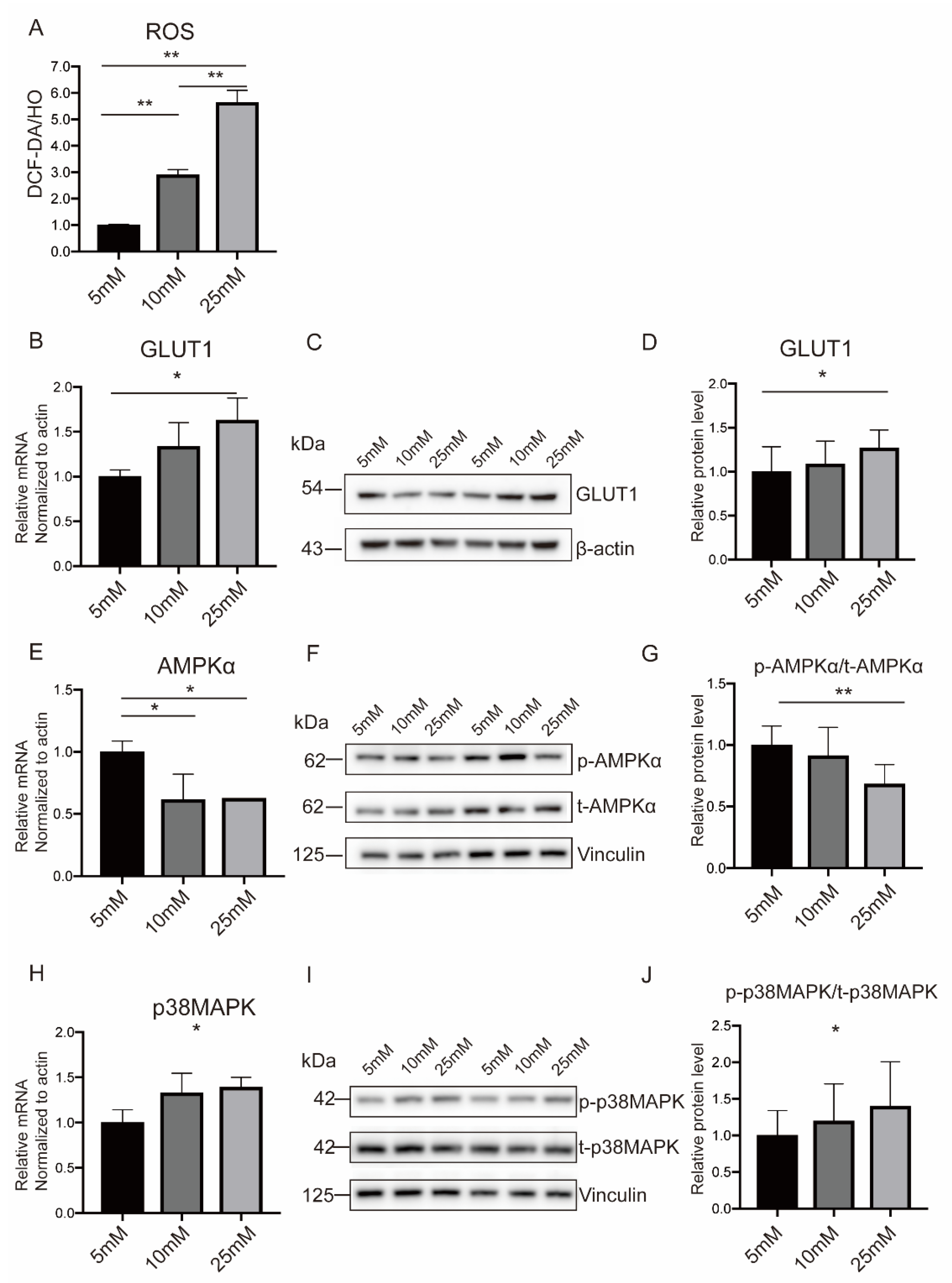
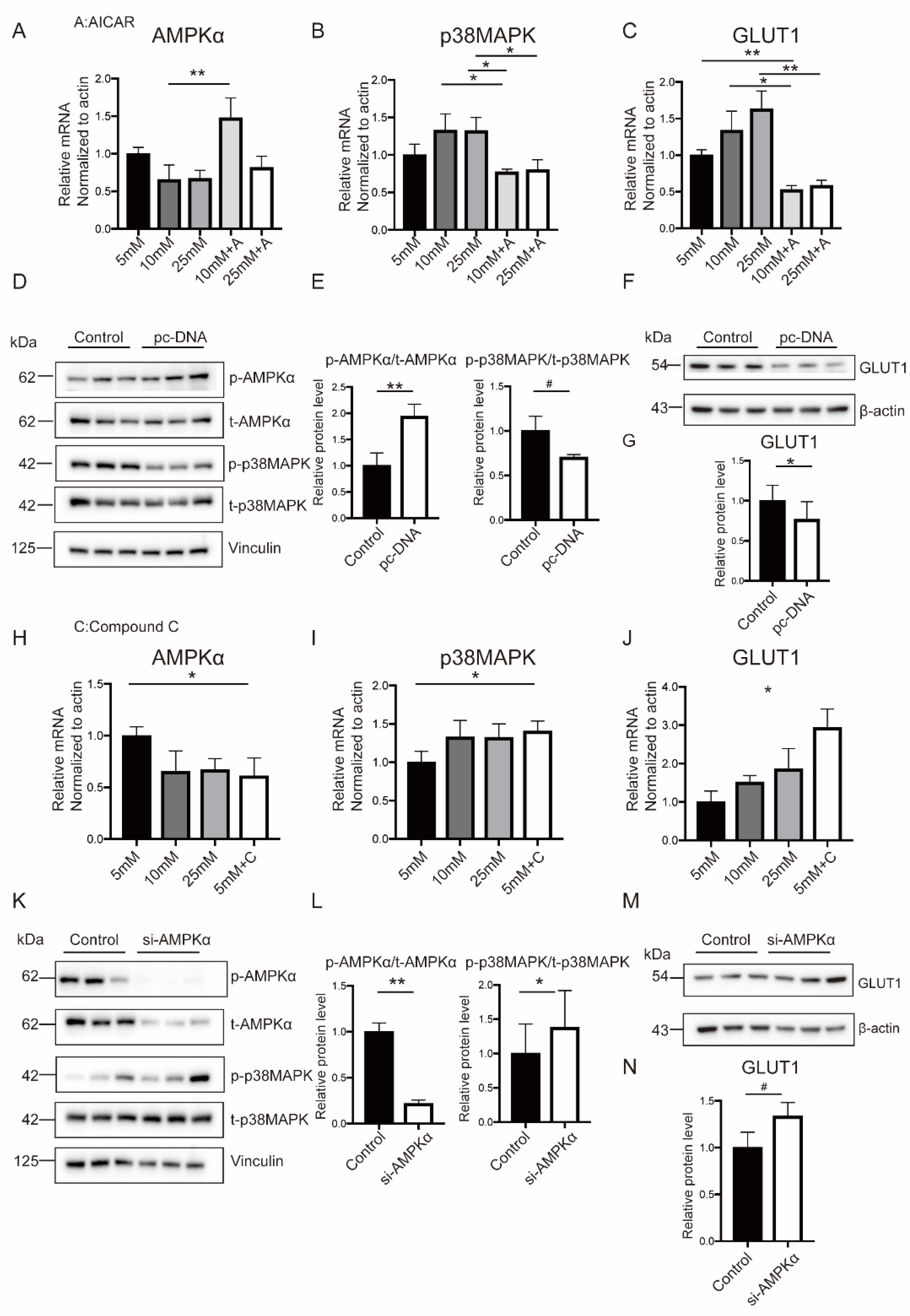
2.4. Hyperglycemia Inhibited AMPKα Activation and Induced p38MAPK Phosphorylation in Both Placental Tissues and In Vitro Trophoblasts
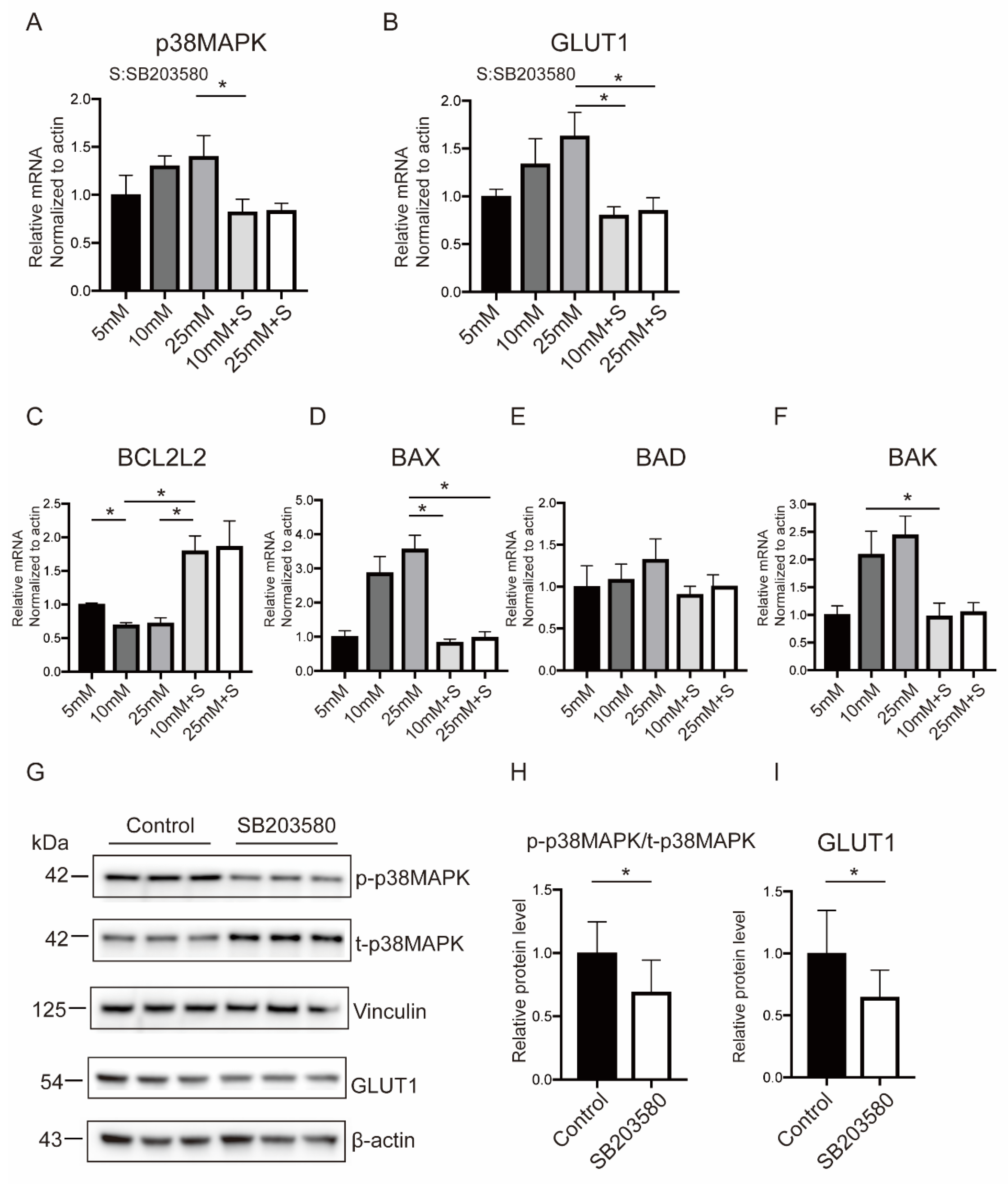
2.5. p38MAPK Mediated Hyperglycemia-Stimulated GLUT1 Expression and Apoptosis in BeWo Cells
2.6. Hyperglycemia-Related OS Augment Activated p38MAPK Pathway through AMPKα
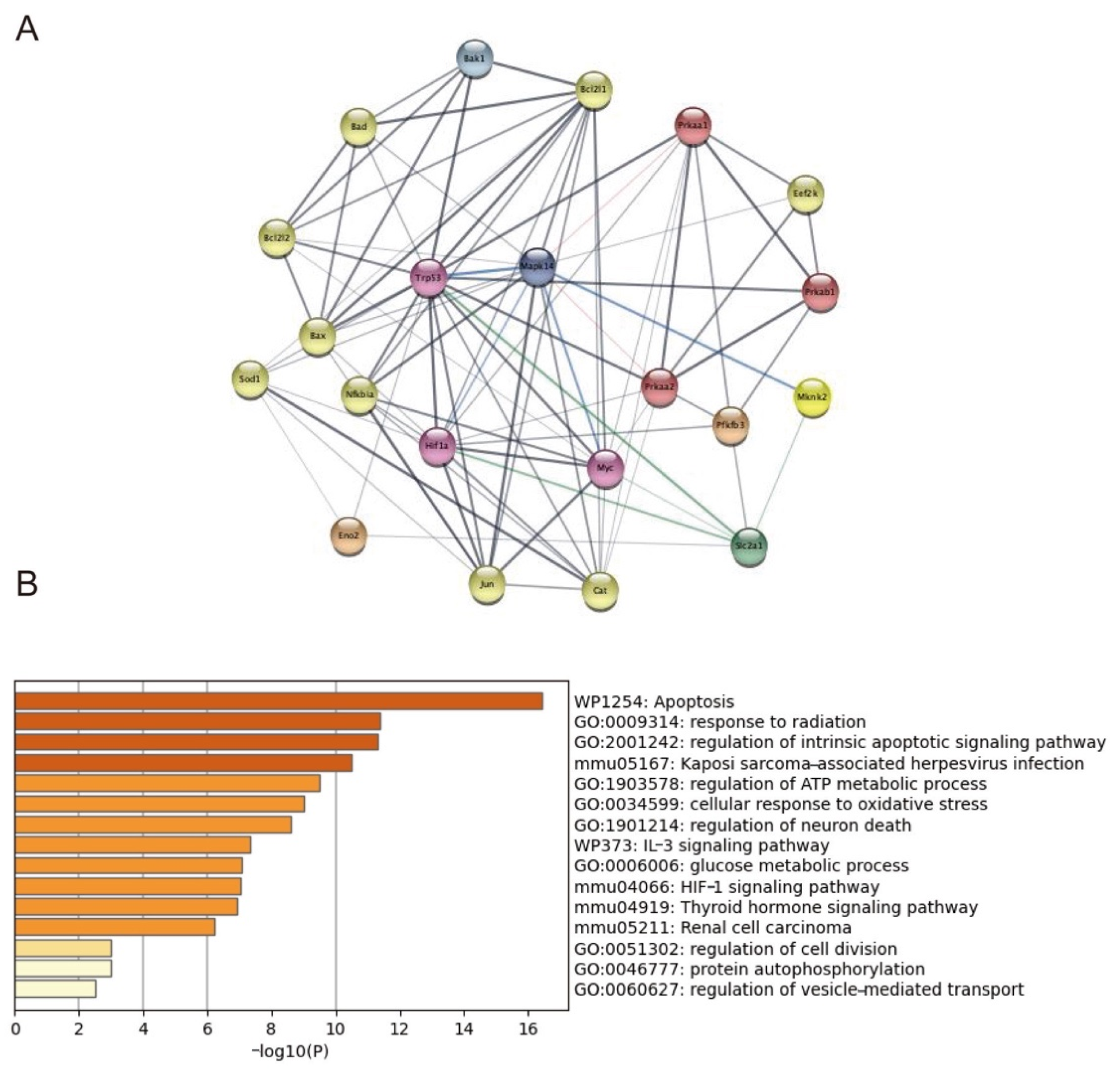
2.7. Trp53, Mknk2, Myc, and HIF1-α Targeted on p38MAPK Involving in GLUT1 Regulation
3. Discussion
4. Materials and Methods
4.1. Study Participants and Sample Collection
4.2. Placental Explant Culture
4.3. Determination of Oxidative Stress Markers
4.4. Measurement of Glycolytic Metabolites by LC-MS/MS
4.5. Cell Culture and Treatments
4.6. Measurement of Intracellular ROS
4.7. Transfection in BeWo Cells
4.8. RNA Isolation and Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
4.9. Western Blot Analysis
4.10. Protein–Protein Interaction Network and Functional Enrichment Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abell, S.K.; De Courten, B.; Boyle, J.A.; Teede, H.J. Inflammatory and other biomarkers: Role in pathophysiology and prediction of gestational diabetes mellitus. Int. J. Mol. Sci. 2015, 16, 13442–13473. [Google Scholar] [CrossRef]
- McElwain, C.J.; Tuboly, E.; McCarthy, F.P.; McCarthy, C.M. Mechanisms of endothelial dysfunction in pre-eclampsia and gestational diabetes mellitus: Windows into future cardiometabolic health? Front. Endocrinol. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Mitton, A.; Permezel, M. In response to oxidative stress, the expression of inflammatory cytokines and antioxidant enzymes are impaired in placenta, but not adipose tissue, of women with gestational diabetes. J. Endocrinol. 2010, 204, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.A.; Imes, S.I.; Liu, D.; McManus, R.; Finegood, D.T.; Polonsky, K.S.; Sturis, J. Defects in insulin secretion and action in women with a history of gestational diabetes. Diabetes 1995, 44, 506–512. [Google Scholar] [CrossRef]
- Dabelea, D.; Crume, T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes 2011, 60, 1849–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalhan, S.C.; D’Angelo, L.J.; Savin, S.M.; Adam, P.A.J. Glucose production in pregnant women at term gestation. Sources of glucose for human fetus. J. Clin. Investig. 1979, 63, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Leonce, J.; Brockton, N.; Robinson, S.; Venkatesan, S.; Bannister, P.; Raman, V.; Murphy, K.; Parker, K.; Pavitt, D.; Teoh, T. Glucose production in the human placenta. Placenta 2006, 27 (Suppl. A), S103–S108. [Google Scholar] [CrossRef]
- Gaither, K.; Quraishi, A.N.; Illsley, N.P. Diabetes alters the expression and activity of the human placental GLUT1 glucose transporter. J. Clin. Endocrinol. Metab. 1999, 84, 695–701. [Google Scholar] [CrossRef]
- Jansson, T.; Ekstrand, Y.; Wennergren, M.; Powell, T.L. Placental glucose transport in gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2001, 184, 111–116. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Pyzlak, M.; Abdalla, N.; Sawicki, W.; Cendrowski, K. Impact of pre-gestational and gestational diabetes mellitus on the expression of glucose transporters GLUT-1, GLUT-4 and GLUT-9 in human term placenta. Endocrine 2017, 55, 799–808. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Pazura-Turowska, M.; Sawicki, W.; Cendrowski, K. Placental expression of glucose transporter proteins in pregnancies complicated by gestational and pregestational diabetes mellitus. Can. J. Diabetes 2018, 42, 209–217. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Majewska, A.; Wątroba, M.; Pyzlak, M.; Bomba-Opoń, D.; Wielgoś, M. Placental expression of glucose transporters GLUT-1, GLUT-3, GLUT-8 and GLUT-12 in pregnancies complicated by gestational and type 1 diabetes mellitus. J. Diabetes Investig. 2021, 13, 560–570. [Google Scholar] [CrossRef]
- Castillo-Castrejon, M.; Yamaguchi, K.; Rodel, R.L.; Erickson, K.; Kramer, A.; Hirsch, N.M.; Rolloff, K.; Jansson, T.; Barbour, L.A.; Powell, T.L. Effect of type 2 diabetes mellitus on placental expression and activity of nutrient transporters and their association with birth weight and neonatal adiposity. Mol. Cell. Endocrinol. 2021, 532, 111319. [Google Scholar] [CrossRef]
- Illsley, N.P. Glucose transporters in the human placenta. Placenta 2000, 21, 14–22. [Google Scholar] [CrossRef]
- Tozour, J.; Hughes, F.; Carrier, A.; Vieau, D.; Delahaye, F. Prenatal hyperglycemia exposure and cellular stress, a sugar-coated view of early programming of metabolic diseases. Biomolecules 2020, 10, 1359. [Google Scholar] [CrossRef] [PubMed]
- Liong, S.; Lappas, M. Activation of AMPK improves inflammation and insulin resistance in adipose tissue and skeletal muscle from pregnant women. J. Physiol. Biochem. 2015, 71, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Itakura, A.; Koya, D.; Kanasaki, K. AMP-activated protein (AMPK) in pathophysiology of pregnancy complications. Int. J. Mol. Sci. 2018, 19, 3076. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yu, X.; Wu, Y.; Fu, H.; Xu, P.; Zheng, Y.; Wen, L.; Yang, X.; Zhang, F.; Hu, M.; et al. Gestational diabetes mellitus-associated hyperglycemia impairs glucose transporter 3 trafficking in trophoblasts through the downregulation of AMP-activated protein kinase. Front. Cell Dev. Biol. 2021, 9, 722024. [Google Scholar] [CrossRef]
- Witczak, C.A.; Sharoff, C.G.; Goodyear, L.J. AMP-activated protein kinase in skeletal muscle: From structure and localization to its role as a master regulator of cellular metabolism. Cell Mol. Life Sci. 2008, 65, 3737–3755. [Google Scholar] [CrossRef]
- Koistinen, H.A.; Galuska, D.; Chibalin, A.V.; Yang, J.; Zierath, J.R.; Holman, G.D.; Wallberg-Henriksson, H. 5-amino-imidazole carboxamide riboside increases glucose transport and cell-surface GLUT4 content in skeletal muscle from subjects with type 2 diabetes. Diabetes 2003, 52, 1066–1072. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.C.; Hucker, K.A.; Holloszy, J.O.; Han, D.H. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes 2004, 53, 330–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, G.; Zhang, Y.; Wang, D.; Yang, R.; Sang, H.; Han, L.; Zhu, Y.; Lu, Y.; Tan, Y.; Shang, Z. GDM-induced macrosomia is reversed by Cav-1 via AMPK-mediated fatty acid transport and GLUT1-mediated glucose transport in placenta. PLoS ONE 2017, 12, e0170490. [Google Scholar] [CrossRef] [PubMed]
- Vila-Bedmar, R.; Lorenzo, M.; Fernández-Veledo, S. Adenosine 5’-monophosphate-activated protein kinase-mammalian target of rapamycin cross talk regulates brown adipocyte differentiation. Endocrinology 2010, 151, 980–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, T.; Murtaza, G.; Metwally, E.; Kalhoro, D.H.; Kalhoro, M.S.; Rahu, B.A.; Sahito, R.G.A.; Yin, Y.; Yang, H.; Chughtai, M.I.; et al. The role of oxidative stress and antioxidant balance in pregnancy. Mediat. Inflamm. 2021, 2021, 9962860. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.A.; Moylan, J.S.; Smith, J.D.; Goodyear, L.J.; Reid, M.B. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J. Physiol. 2009, 587, 3363–3373. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Zhang, L.; Gao, P.; Wu, X. Adiponectin attenuates high glucose-induced apoptosis through the AMPK/p38 MAPK signaling pathway in NRK-52E cells. PLoS ONE 2017, 12, e0178215. [Google Scholar]
- Jing, Y.; Liu, W.; Cao, H.; Zhang, D.; Yao, X.; Zhang, S.; Xia, H.; Li, D.; Wang, Y.-C.; Yan, J.; et al. Hepatic p38α regulates gluconeogenesis by suppressing AMPK. J. Hepatol. 2015, 62, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Jauniaux, E.; Murray, A.J. Oxygen and placental development; parallels and differences with tumour biology. Placenta 2017, 56, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Desoye, G.; Hauguel-de Mouzon, S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care 2007, 30 (Suppl. 2), S120–S126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, Y.C.; Zhang, Z.; Barila, G.; Green-Brown, A.; Elovitz, M.A.; Simmons, R.A. Intrauterine inflammation alters the transcriptome and metabolome in placenta. Front. Physiol. 2020, 11, 592689. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngo, C.; Jayabalan, N.; Salomon, C.; Lappas, M. Molecular pathways disrupted by gestational diabetes mellitus. J. Mol. Endocrinol. 2019, 63, R51–R72. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cervantes, M.; Peña-Montes, D.J.; Montoya-Pérez, R.; Trujillo, X.; Huerta, M.; López-Vázquez, M.Á.; Olvera-Cortés, M.E.; Saavedra-Molina, A. Gestational diabetes triggers oxidative stress in hippocampus and cerebral cortex and cognitive behavior modifications in rat offspring: Age- and sex-dependent effects. Nutrients 2020, 12, 376. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.J.; Vanderpeet, C.L.; Bartho, L.A.; McKeating, D.R.; Cuffe, J.S.; Holland, O.J.; Perkins, A.V. Mitochondrial dysfunction in placental trophoblast cells experiencing gestational diabetes mellitus. J. Physiol. 2021, 599, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, Y.; Chen, R.; Wei, Y.; Feng, Y.; Zheng, W.; Liao, H.; Zhang, Z. Aberrant expression of oxidative stress related proteins affects the pregnancy outcome of gestational diabetes mellitus patients. Am. J. Transl. Res. 2019, 11, 269–279. [Google Scholar] [PubMed]
- Sgarbosa, F.; Barbisan, L.F.; Brasil, M.A.; Costa, E.; Calderon, I.; Gonçalves, C.R.; Bevilacqua, E.; Rudge, M.V. Changes in apoptosis and Bcl-2 expression in human hyperglycemic, term placental trophoblast. Diabetes Res. Clin. Pract. 2006, 73, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Emiliano, J.; Fajardo-Araujo, M.E.; Zúñiga-Trujillo, I.; Pérez-Vázquez, V.; Sandoval-Salazar, C.; Órnelas-Vázquez, J.K. Mitochondrial content, oxidative, and nitrosative stress in human full-term placentas with gestational diabetes mellitus. Reprod. Biol. Endocrinol. 2017, 15, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziętek, M.; Celewicz, Z.; Szczuko, M. Short-chain fatty acids, maternal microbiota and metabolism in pregnancy. Nutrients 2021, 13, 1244. [Google Scholar] [CrossRef] [PubMed]
- Jayabalan, N.; Lai, A.; Ormazabal, V.; Adam, S.; Guanzon, D.; Palma, C.; Scholz-Romero, K.; Lim, R.; Jansson, T.; McIntyre, H.D.; et al. Adipose tissue exosomal proteomic profile reveals a role on placenta glucose metabolism in gestational diabetes mellitus. J. Clin. Endocrinol. Metab. 2019, 104, 1735–1752. [Google Scholar] [CrossRef]
- Valent, A.M.; Choi, H.; Kolahi, K.S.; Thornburg, K.L. Hyperglycemia and gestational diabetes suppress placental glycolysis and mitochondrial function and alter lipid processing. FASEB J. 2021, 35, e21423. [Google Scholar] [CrossRef]
- Castillo-Castrejon, M.; Powell, T.L. Placental nutrient transport in gestational diabetic pregnancies. Front. Endocrinol. 2017, 8, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanirowski, P.J.; Lipa, M.; Bomba-Opoń, D.; Wielgoś, M. Expression of placental glucose transporter proteins in pregnancies complicated by fetal growth disorders. Adv. Protein Chem. Struct. Biol. 2021, 123, 95–131. [Google Scholar] [PubMed]
- Baumann, M.U.; Deborde, S.; Illsley, N.P. Placental glucose transfer and fetal growth. Endocrine 2002, 19, 13–22. [Google Scholar] [CrossRef]
- Araújo, J.R.; Keating, E.; Martel, F. Impact of gestational diabetes mellitus in the maternal-to-fetal transport of nutrients. Curr. Diabetes Rep. 2015, 15, 569. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Pyzlak, M.; Abdalla, N.; Sawicki, W.; Cendrowski, K. Analysis of correlations between the placental expression of glucose transporters GLUT-1, GLUT-4 and GLUT-9 and selected maternal and fetal parameters in pregnancies complicated by diabetes mellitus. J. Matern. Fetal Neonatal Med. 2019, 32, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Vajnerova, O.; Kafka, P.; Kratzerova, T.; Chalupsky, K.; Hampl, V. Pregestational diabetes increases fetoplacental vascular resistance in rats. Placenta 2018, 63, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Starikov, R.; Inman, K.; Chen, K.; Lopes, V.; Coviello, E.; Pinar, H.; He, M. Comparison of placental findings in type 1 and type 2 diabetic pregnancies. Placenta 2014, 35, 1001–1006. [Google Scholar] [CrossRef]
- López-Tinoco, C.; Jiménez-Blázquez, J.L.; Larrán-Escandón, L.; Roca-Rodríguez, M.D.M.; Bugatto, F.; Diosdado, M.A. Effect of different insulin therapies on obstetric-fetal outcomes. Sci. Rep. 2019, 9, 17650. [Google Scholar] [CrossRef]
- Hulme, C.H.; Stevens, A.; Dunn, W.; Heazell, A.E.P.; Hollywood, K.; Begley, P.; Westwood, M.; Myers, J.E. Identification of the functional pathways altered by placental cell exposure to high glucose: Lessons from the transcript and metabolite interactome. Sci. Rep. 2018, 8, 5270. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Gupta, A.; Sehgal, A.; Sharma, S.; Singh, S.; Sharma, N.; Diaconu, C.C.; Rahdar, A.; Hafeez, A.; Bhatia, S.; et al. A spotlight on underlying the mechanism of AMPK in diabetes complications. Inflamm. Res. 2021, 70, 939–957. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Rotariu, D.; Babes, E.E.; Tit, D.M.; Moisi, M.; Bustea, C.; Stoicescu, M.; Radu, A.F.; Vesa, C.M.; Behl, T.; Bungau, A.F.; et al. Oxidative stress—Complex pathological issues concerning the hallmark of cardiovascular and metabolic disorders. Biomed. Pharmacother. 2022, 152, 113238. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Wu, C.P.; Chen, S.F. Differential changes in Akt and AMPK phosphorylation regulating mTOR activity in the placentas of pregnancies complicated by fetal growth restriction and gestational diabetes mellitus with large-for-gestational age infants. Front. Med. 2021, 8, 788969. [Google Scholar] [CrossRef]
- Nomoto, H.; Pei, L.; Montemurro, C.; Rosenberger, M.; Furterer, A.; Coppola, G.; Nadel, B.; Pellegrini, M.; Gurlo, T.; Butler, P.C.; et al. Activation of the HIF1α/PFKFB3 stress response pathway in beta cells in type 1 diabetes. Diabetologia 2020, 63, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Duran, J.; Obach, M.; Navarro-Sabate, A.; Manzano, A.; Gómez, M.; Rosa, J.L.; Ventura, F.; Perales, J.C.; Bartrons, R. Pfkfb3 is transcriptionally upregulated in diabetic mouse liver through proliferative signals. FEBS J. 2009, 276, 4555–4568. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, W.; Zhong, Y.; Li, Q.; Wu, M.; Yang, L.; Liu, X.; Zou, L. Metformin corrects glucose metabolism reprogramming and NLRP3 inflammasome-induced pyroptosis via inhibiting the TLR4/NF-κB/PFKFB3 signaling in trophoblasts: Implication for a potential therapy of preeclampsia. Oxid. Med. Cell. Longev. 2021, 2021, 1806344. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, J.; Liang, H.; Cai, Y.; Li, X.; Yan, L.; Zhou, L.; Shan, L.; Wang, H. Human umbilical cord-derived mesenchymal stem cells not only ameliorate blood glucose but also protect vascular endothelium from diabetic damage through a paracrine mechanism mediated by MAPK/ERK signaling. Stem Cell Res. Ther. 2022, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; He, Z.; Wu, Y.; Zeng, C.; Zheng, Z.; Zhang, H.; Lv, C.; Yuan, Y.; Wu, H.; Ye, J.; et al. Instant dark tea alleviates hyperlipidaemia in high-fat diet-fed rat: From molecular evidence to redox balance and beyond. Front. Nutr. 2022, 9, 819980. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.Y.; Lee, Y.K.; Kim, J.E.; Nam, S.H.; Goo, J.S.; Choi, S.I.; Choi, Y.H.; Bae, C.J.; Woo, J.M.; Cho, J.S. Differential regulation of the biosynthesis of glucose transporters by the PI3-K and MAPK pathways of insulin signaling by treatment with novel compounds from Liriope platyphylla. Int. J. Mol. Med. 2011, 27, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Wang, H.; Li, J.-J.; Zhang, Y.-L.; Xin, L.; Li, F.; Lou, S.-J. The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neuronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain Behav. Immun. 2016, 57, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Hu, J.Y.; Liu, B.B.; Li, J.J.; Li, F.; Lou, S. The molecular mechanisms of excessive hippocampal endoplasmic reticulum stress depressing cognition-related proteins expression and the regulatory effects of Nrf2. Neuroscience 2020, 431, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.F.; Galardo, M.N.; Pellizzari, E.H.; Meroni, S.B.; Cigorraga, S.B. Molecular mechanisms involved in Sertoli cell adaptation to glucose deprivation. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E907–E914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, O.; Ramirez, V.I.; Lager, S.; Gaccioli, F.; Dudley, D.J.; Powell, T.; Jansson, T. Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am. J. Obstet. Gynecol. 2015, 212, e221–e227. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 14) | GDM1 (n = 10) | GDM2 (n = 9) | PGDM (n = 10) | p-Value | |
|---|---|---|---|---|---|
| Age (years) | 32.78 ± 3.577 | 33.70 ± 4.448 | 35.67 ± 3.605 | 34.70 ± 4.877 | 0.4 |
| Gestational age (Weeks) | 38.64 ± 0.745 | 38.80 ± 0.632 | 38.89 ± 0.333 | 38.60 ± 0.516 | 0.681 |
| p-BMI (kg/m2) | 21.68 ± 1.765 | 22.27 ± 2.491 | 26.56 ± 3.092 | 25.77 ± 4.803 | 0.001 ** |
| GWG (kg) | 13.37 ± 4.239 | 11.40 ± 2.989 | 9.49 ± 2.875 | 8.45 ± 3.218 | 0.008 ** |
| GLU0 (mmol/L) a | 4.51 ± 0.180 | 5.17 ± 0.415 | 5.53 ± 0.420 | - | <0.0001 **** |
| GLU1 (mmol/L) a | 7.88 ± 1.161 | 10.23 ± 1.335 | 10.00 ± 1.321 | - | <0.0001 **** |
| GLU2 (mmol/L) a | 6.57 ± 0.873 | 9.21 ± 1.086 | 8.15 ± 1.520 | - | <0.0001 **** |
| AUC | 13.42 ± 1.495 | 17.42 ± 1.402 | 16.84 ± 1.952 | - | <0.0001 **** |
| Third trimester glucose (mmol/L) | 4.48 ± 0.371 | 4.71 ± 0.463 | 5.17 ± 0.925 | 5.19 ± 0.838 | 0.035 * |
| Fetal birth weight (g) | 3439.28 ± 330.657 | 3580.00 ± 512.809 | 3582.78 ± 256.065 | 3350.00 ± 324.414 | 0.509 |
| Height (cm) | 50.21 ± 0.975 | 50.50 ± 1.581 | 50.33 ± 1.000 | 50.28 ± 1.054 | 0.858 |
| Ponderal Index (kg/m3) | 2.91 ± 0.143 | 2.73 ± 0.260 | 2.81 ± 0.133 | 2.66 ± 0.200 | 0.393 |
| Head circumference (cm) | 33.96 ± 0.499 | 34.07 ± 0.861 | 34.39 ± 0.928 | 34.11 ± 0.782 | 0.626 |
| Placenta weight (g) | 580.00 ± 80.288 | 641.33 ± 158.338 | 602.22 ± 69.061 | 630.00 ± 92.736 | 0.509 |
| Placenta volume (cm3) | 704.86 ± 71.471 | 778.00 ± 90.985 | 954.89 ± 198.515 | 870.30 ± 158.711 | <0.0001 **** |
| Placental coefficient | 0.169 ± 0.023 | 0.186 ± 0.068 | 0.169 ± 0.025 | 0.192 ± 0.030 | 0.456 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Ning, J.; Huai, J.; Yang, H. Hyperglycemia in Pregnancy-Associated Oxidative Stress Augments Altered Placental Glucose Transporter 1 Trafficking via AMPKα/p38MAPK Signaling Cascade. Int. J. Mol. Sci. 2022, 23, 8572. https://doi.org/10.3390/ijms23158572
Wang S, Ning J, Huai J, Yang H. Hyperglycemia in Pregnancy-Associated Oxidative Stress Augments Altered Placental Glucose Transporter 1 Trafficking via AMPKα/p38MAPK Signaling Cascade. International Journal of Molecular Sciences. 2022; 23(15):8572. https://doi.org/10.3390/ijms23158572
Chicago/Turabian StyleWang, Shuxian, Jie Ning, Jing Huai, and Huixia Yang. 2022. "Hyperglycemia in Pregnancy-Associated Oxidative Stress Augments Altered Placental Glucose Transporter 1 Trafficking via AMPKα/p38MAPK Signaling Cascade" International Journal of Molecular Sciences 23, no. 15: 8572. https://doi.org/10.3390/ijms23158572
APA StyleWang, S., Ning, J., Huai, J., & Yang, H. (2022). Hyperglycemia in Pregnancy-Associated Oxidative Stress Augments Altered Placental Glucose Transporter 1 Trafficking via AMPKα/p38MAPK Signaling Cascade. International Journal of Molecular Sciences, 23(15), 8572. https://doi.org/10.3390/ijms23158572