
ACBD3 Bioinformatic Analysis and Protein Expression in Breast Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ACBD3 Expression in Tumours and Normal Tissue
2.2. ACBD3 Gene Amplification and Mutation in Cancer
2.3. Copy Number Variation and Promoter Methylation of ACBD3 in Breast Cancer
2.4. ACBD3 Transcription Factors in Breast Tissue
2.5. ACBD3 mRNA Expression and Breast Cancer Patient Prognosis
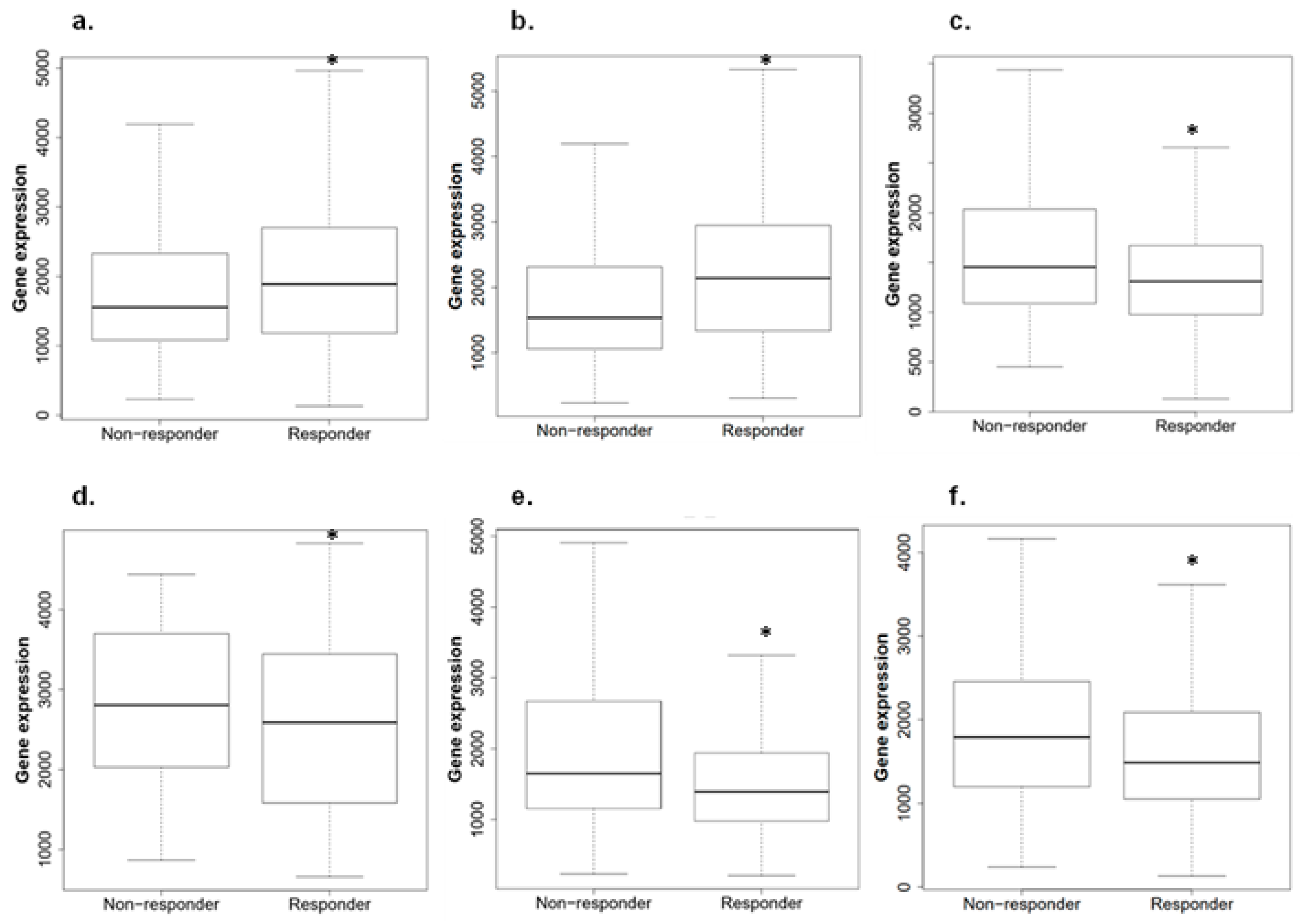
2.6. ACBD3 Expression in Responders and Non-Responders to Chemotherapy in Breast Cancer
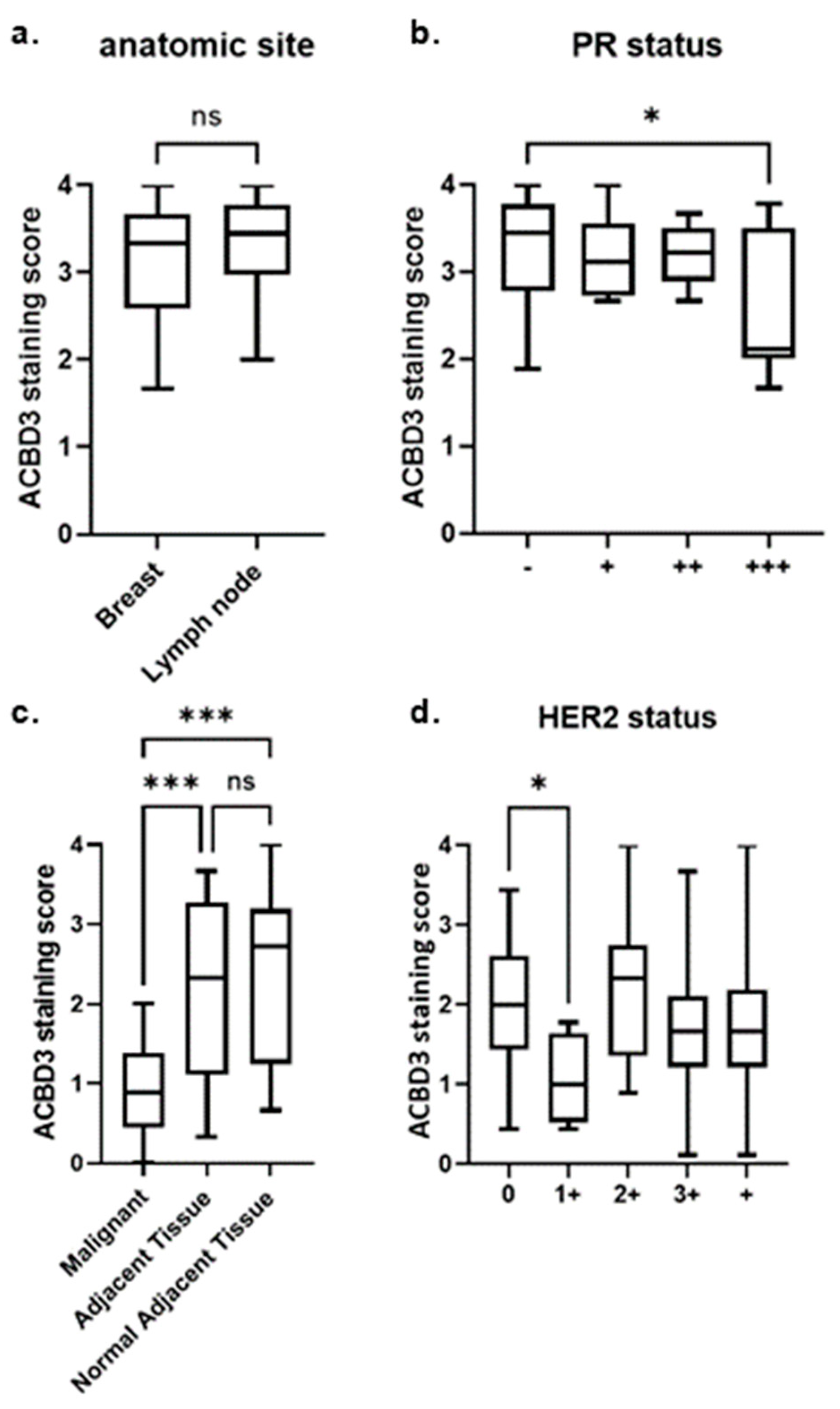
2.7. Aberrant Protein Expression of ACBD3 in Breast Cancer Cores
2.7.1. ACBD3 Protein Expression in Malignant Breast Tissue and Metastatic Lymph Node Tissue
2.7.2. ACBD3 Protein Expression Is Lower in Malignant Breast Tissue Compared to Cancer Adjacent and Normal Adjacent Breast Tissue
2.7.3. ACBD3 Protein Expression in Malignant Breast Tissue of Multiple Subtype Receptor Status and Pathology
3. Discussion
4. Materials and Methods
4.1. Bioinformatic Analysis
4.2. Immunohistochemistry
4.3. Core Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, J.; Liu, J.; Culty, M.; Papadopoulos, V. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): An emerging signalling molecule. Prog. Lipid Res. 2010, 49, 218–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, Y.; Ma, Y.; Yeh, M.; Yin, H.; Li, Z.; Yeh, K.Y.; Glass, J. DMT1 (IRE) expression in intestinal and erythroid cells is regulated by peripheral benzodiazepine receptor-associated protein 7. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 302, G1180–G1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohda, M.; Misumi, Y.; Yamamoto, A.; Yano, A.; Nakamura, N.; Ikehara, Y. Identification and characterization of a novel Golgi protein, GCP60, that interacts with the integral membrane protein giantin. J. Biol. Chem. 2001, 276, 45298–45306. [Google Scholar] [CrossRef] [Green Version]
- Belman, J.P.; Bian, R.R.; Habtemichael, E.N.; Li, D.T.; Jurczak, M.J.; Alcázar-Román, A.; McNally, L.J.; Shulman, G.I.; Bogan, J.S. Acetylation of TUG protein promotes the accumulation of GLUT4 glucose transporters in an insulin-responsive intracellular compartment. J. Biol. Chem. 2015, 290, 4447–4463. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Hicks, S.W.; Simon, D.; Machamer, C.E. GCP60 preferentially interacts with a caspase-generated golgin-160 fragment. J. Biol. Chem. 2006, 281, 27924–27931. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Machamer, C.E. Identification of a redox-sensitive cysteine in GCP60 that regulates its interaction with golgin-160. J. Biol. Chem. 2007, 282, 29874–29881. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Bao, M.; Christiano, R.; Li, S.; Mei, J.; Zhu, L.; Mao, F.; Yue, Q.; Zhang, P.; Jing, S.; et al. ACBD3 functions as a scaffold to organize the Golgi stacking proteins and a Rab33b-GAP. FEBS Lett. 2017, 591, 2793–2802. [Google Scholar] [CrossRef]
- Shinoda, Y.; Fujita, K.; Saito, S.; Matsui, H.; Kanto, Y.; Nagaura, Y.; Fukunaga, K.; Tamura, S.; Kobayashi, T. Acyl-CoA binding domain containing 3 (ACBD3) recruits the protein phosphatase PPM1L to ER-Golgi membrane contact sites. FEBS Lett. 2012, 586, 3024–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klima, M.; Tóth, D.J.; Hexnerova, R.; Baumlova, A.; Chalupska, D.; Tykvart, J.; Rezabkova, L.; Sengupta, N.; Man, P.; Dubankova, A.; et al. Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Sci. Rep. 2016, 6, 23641. [Google Scholar] [CrossRef]
- Govers, R. Cellular regulation of glucose uptake by glucose transporter GLUT4. Adv. Clin. Chem. 2014, 66, 173–240. [Google Scholar]
- Garrido, P.; Osorio, F.G.; Morán, J.; Cabello, E.; Alonso, A.; Freije, J.M.; González, C. Loss of GLUT4 induces metabolic reprogramming and impairs viability of breast cancer cells. J. Cell Physiol. 2015, 230, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; LaPara, K.; Yee, D. Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells. Oncogene 2016, 35, 4235–4243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Degenhardt, B.; Tobin, D.; Yao, Z.; Tasken, K.; Papadopoulos, V. Identification, localization, and function in steroidogenesis of PAP7: A peripheral-type benzodiazepine receptor-and PKA (RIα)-associated protein. Mol. Endocrinol. 2001, 15, 2211–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, K.E.; Papadopoulos, V. Peripheral-type benzodiazepine receptors mediate translocation of cholesterol from outer to inner mitochondrial membranes in adrenocortical cells. J. Biol. Chem. 1990, 265, 15015–15022. [Google Scholar] [CrossRef]
- Mantovani, G.; Bondioni, S.; Lania, A.G.; Rodolfo, M.; Peverelli, E.; Polentarutti, N.; Veliz Rodriguez, T.; Ferrero, S.; Bosari, S.; Beck-Peccoz, P.; et al. High expression of PKA regulatory subunit 1A protein is related to proliferation of human melanoma cells. Oncogene 2008, 27, 1834–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDaid, H.M.; Cairns, M.T.; Atkinson, R.J.; McAleer, S.; Harkin, D.P.; Gilmore, P.; Johnston, P. Increased expression of the RIalpha subunit of the cAMP-dependent protein kinase A is associated with advanced stage ovarian cancer. Br. J. Cancer 1999, 79, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Atkins, J.B.; Rompani, S.B.; Bancescu, D.L.; Petersen, P.H.; Tang, H.; Zou, K.; Stewart, S.B.; Zhong, W. The mammalian Golgi regulates numb signaling in asymmetric cell division by releasing ACBD3 during mitosis. Cell 2007, 129, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [Green Version]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006, 66, 1517. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shao, X.; Sun, H.; Liu, K.; Ding, Z.; Chen, J.; Fang, L.; Su, W.; Hong, Y.; Li, H.; et al. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 2016, 7, 61036–61053. [Google Scholar] [CrossRef] [Green Version]
- García-Heredia, J.M.; Sivianes, E.M.V.; Lucena-Cacace, A.; Molina-Pinelo, S.; Carnero, A. Numb-like (NumbL) downregulation increases tumorigenicity, cancer stem cell-like properties and resistance to chemotherapy. Oncotarget 2016, 7, 63611–63628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yang, L.; Pei, Y.Y.; Wang, J.; Wu, H.; Yuan, J.; Wang, L. Overexpressed ACBD3 has prognostic value in human breast cancer and promotes the self-renewal potential of breast cancer cells by activating the Wnt/beta-catenin signaling pathway. Exp. Cell Res. 2018, 363, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Qian, Y.; Gim, B.; Lee, I. Acyl-CoA-binding domain-containing 3 (ACBD3; PAP7; GCP60): A multi-functional membrane domain organizer. Int. J. Mol. Sci. 2019, 20, 2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton-Gisby, J.; Harvey, A.J. ACBD3, its cellular interactors, and its role in breast cancer. Cancer Stud. Ther. 2020, 5, 1–7. [Google Scholar]
- Rostoker, R.; Abelson, S.; Bitton-Worms, K.; Genkin, I.; Ben-Shmuel, S.; Dakwar, M.; Orr, Z.S.; Caspi, A.; Tzukerman, M.; LeRoith, D. Highly specific role of the insulin receptor in breast cancer progression. Endocr.-Relat. Cancer 2015, 22, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Morrow, A.A.; Alipour, M.A.; Bridges, D.; Yao, Z.; Saltiel, A.R.; Lee, J.M. The lipid kinase PI4KIIIβ is highly expressed in breast tumors and activates Akt in cooperation with Rab11a. Mol. Cancer Res. 2014, 12, 1492. [Google Scholar] [CrossRef] [Green Version]
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Di Fiore, P.P. NUMB controls p53 tumour suppressor activity. Nature 2008, 451, 76–80. [Google Scholar] [CrossRef]
- Acharya, S.; Xu, J.; Wang, X.; Jain, S.; Wang, H.; Zhang, Q.; Chang, C.C.; Bower, J.; Arun, B.; Seewaldt, V.; et al. Downregulation of GLUT4 contributes to effective intervention of estrogen receptor-negative/HER2-overexpressing early stage breast disease progression by lapatinib. Am. J. Cancer Res. 2016, 6, 981–995. [Google Scholar]
- Hanahan, D.; Weingberg, R. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Matyakhina, L.; Han, Z.; Sandrini, F.; Bei, T.; Stratakis, C.A.; Papadopoulos, V. Molecular cloning, chromosomal localization of human peripheral-type benzodiazepine receptor and PKA regulatory subunit type 1A (PRKAR1A)-associated protein PAP7, and studies in PRKAR1A mutant cells and tissues. FASEB J. 2003, 17, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Arakane, F.; King, S.R.; Du, Y.; Kallen, C.B.; Walsh, L.P.; Watari, H.; Stocco, D.M.; Strauss, J.F. Phosphorylation of steroidogenic acute regulatory protein (StAR) modulates its steroidogenic activity. J. Biol. Chem. 1997, 272, 32656–32662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatliff, J.; East, D.A.; Singh, A.; Alvarez, M.S.; Frison, M.; Matic, I.; Ferraina, C.; Sampson, N.; Turkheimer, F.; Campanella, M. A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis. 2017, 8, e2896. [Google Scholar] [CrossRef] [Green Version]
- Orsetti, B.; Nugoli, M.; Cervera, N.; Lasorsa, L.; Chuchana, P.; Rougé, C.; Ursule, L.; Nguyen, C.; Bibeau, F.; Rodriguez, C.; et al. Genetic profiling of chromosome 1 in breast cancer: Mapping of regions of gains and losses and identification of candidate genes on 1q. Br. J. Cancer 2006, 95, 1439. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Zhang, M.; Shang, Z.; Li, J.; Zhang, S.; Lian, D.; Zhang, R. Genome-wide haplotype association study identify the FGFR2 gene as a risk gene for acute myeloid leukemia. Oncotarget 2017, 8, 7891–7899. [Google Scholar] [CrossRef] [Green Version]
- Ujjainwala, A.L.; Courtney, C.D.; Rhoads, S.G.; Rhodes, J.S.; Christian, C.A. Genetic loss of diazepam binding inhibitor in mice impairs social interest. Genes Brain Behav. 2018, 17, e12442. [Google Scholar] [CrossRef]
- Lanfray, D.; Caron, A.; Roy, M.C.; Laplante, M.; Morin, F.; Leprince, J.; Tonon, M.C.; Richard, D. Involvement of the Acyl-CoA binding domain containing 7 in the control of food intake and energy expenditure in mice. Elife 2016, 2, 5. [Google Scholar]
- Lanfray, D.; Richard, D. Emerging Signaling pathway in arcuate feeding-related neurons: Role of the Acbd7. Front. Neurosci. 2017, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Smiraglia, D.J.; Plass, C. The study of aberrant methylation in cancer via restriction landmark genomic scanning. Oncogene 2002, 21, 5414–5426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Yang, M.; Park, J.Y. DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol. Biol. 2012, 863, 67–109. [Google Scholar]
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene promoter methylation and cancer: An umbrella review. Gene 2019, 710, 333–340. [Google Scholar] [CrossRef]
- Achinger-Kawecka, J.; Valdes-Mora, F.; Luu, P.L.; Giles, K.A.; Caldon, C.E.; Qu, W.; Nair, S.; Soto, S.; Locke, W.J.; Yeo-Teh, N.S.; et al. Epigenetic reprogramming at estrogen-receptor binding sites alters 3D chromatin landscape in endocrine-resistant breast cancer. Nat. Commun. 2020, 11, 320. [Google Scholar] [CrossRef] [Green Version]
- Ochsner, S.A.; Abraham, D.; Martin, K.; Ding, W.; McOwiti, A.; Kankanamge, W.; Wang, Z.; Andreano, K.; Hamilton, R.A.; Chen, Y.; et al. The Signaling Pathways Project: An integrated ‘omics knowledgebase for mammalian cellular signaling pathways. Sci. Data 2019, 6, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlafstein, A.J.; Withers, A.E.; Rudra, S.; Danelia, D.; Switchenko, J.M.; Mister, D.; Harari, S.; Zhang, H.; Daddacha, W.; Ehdaivand, S.; et al. CDK9 expression shows role as a potential prognostic biomarker in breast cancer patients who fail to achieve pathologic complete response after neoadjuvant chemotherapy. Int. J. Breast Cancer 2018, 2018, 6945129. [Google Scholar] [CrossRef] [Green Version]
- Brisard, D.; Eckerdt, F.; Marsh, L.A.; Blyth, G.T.; Jain, S.; Cristofanilli, M.; Horiuchi, D.; Platanias, L.C. Antineoplastic effects of selective CDK9 inhibition with atuveciclib on cancer stem-like cells in triple-negative breast cancer. Oncotarget 2018, 9, 37305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, R.P.; He, J.; Van Der Noord, V.E.; Redel, J.; Foekens, J.A.; Martens, J.W.; Smid, M.; Zhang, Y.; Van de Water, B. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res. 2019, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Aulmann, S.; Bläker, H.; Penzel, R.; Rieker, R.J.; Otto, H.F.; Sinn, H.P. CTCF gene mutations in invasive ductal breast cancer. Breast Cancer Res. Treat. 2003, 80, 347–352. [Google Scholar] [CrossRef]
- Docquier, F.; Kita, G.X.; Farrar, D.; Jat, P.; O’Hare, M.; Chernukhin, I.; Gretton, S.; Mandal, A.; Alldridge, L.; Klenova, E. Decreased poly(ADP-ribosyl)ation of CTCF, a transcription factor, is associated with breast cancer phenotype and cell proliferation. Clin. Cancer Res. 2009, 15, 5762–5771. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, M.; Lee, J.Y.; Kim, M.H. CTCF negatively regulates HOXA10 expression in breast cancer cells. Biochem. Biophys. Res. Commun. 2015, 467, 828–834. [Google Scholar] [CrossRef]
- Oh, S.; Oh, C.; Yoo, K.H. Functional roles of CTCF in breast cancer. BMB Rep. 2017, 50, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Damaschke, N.A.; Gawdzik, J.; Avilla, M.; Yang, B.; Svaren, J.; Roopra, A.; Luo, J.H.; Yu, Y.P.; Keles, S.; Jarrard, D.F. CTCF loss mediates unique DNA hypermethylation landscapes in human cancers. Clin. Epigenetics 2020, 12, 80. [Google Scholar] [CrossRef]
- Grimm, S.L.; Rosen, J.M. The role of C/EBPβ in mammary gland development and breast cancer. J. Mammary Gland. Biol. Neoplasia 2003, 8, 191–204. [Google Scholar] [CrossRef]
- Zahnow, C.A. CCAAT/enhancer-binding protein beta: Its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev. Mol. Med. 2009, 11, e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzejamska, E.; Johansson, J.; Jirström, K.; Prakash, V.; Ananthaseshan, S.; Boon, L.; Fuxe, J.; Religa, P. C/EBPβ expression is an independent predictor of overall survival in breast cancer patients by MHCII/CD4-dependent mechanism of metastasis formation. Oncogenesis 2014, 3, e125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, A.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef] [PubMed]
- Fekete, J.T.; Győrffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef] [Green Version]
- Roche, S.; Fumagalli, S.; Courtneidge, S.A. Requirement for Src family protein tyrosine kinases in G2 for fibroblast cell division. Science 1995, 269, 1567–1569. [Google Scholar] [CrossRef] [PubMed]
- Resnick, R.J.; Taylor, S.J.; Lin, Q.; Shalloway, D. Phosphorylation of the Src substrate Sam68 by Cdc2 during mitosis. Oncogene 1997, 15, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, S.; Totty, N.F.; Hsuan, J.J.; Courtneidge, S.A. A target for Src in mitosis. Nature 1994, 368, 871–874. [Google Scholar] [CrossRef]
- Taylor, S.J.; Shalloway, D. An RNA-binding protein associated with Src through its SH2 and SH3 domains in mitosis. Nature 1994, 368, 867–871. [Google Scholar] [CrossRef]
- Pfau, R.; Tzatsos, A.; Kampranis, S.C.; Serebrennikova, O.B.; Bear, S.E.; Tsichlis, P.N. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc. Natl. Acad. Sci. USA 2008, 105, 1907–1912. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Gao, Y.; Zhang, Z.; Cao, Q.; Zhang, X.; Zou, J.; Cao, Y. Kdm2a/b lysine demethylases regulate canonical Wnt signaling by modulating the stability of nuclear β-catenin. Dev. Cell 2015, 33, 660–674. [Google Scholar] [CrossRef] [Green Version]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, G.M.; Lozada, K.L.; Miedler, J.D.; Harburg, G.; Hewitt, S.C.; Mosley, J.D.; Godwin, A.K.; Korach, K.S.; Visvader, J.E.; Kaestner, K.H.; et al. FOXA1 is an essential determinant of ERalpha expression and mammary ductal morphogenesis. Development 2010, 137, 2045–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Thorat, M.A.; Marchio, C.; Morimiya, A.; Savage, K.; Nakshatri, H.; Reis-Filho, J.S.; Badve, S. Forkhead box A1 expression in breast cancer is associated with luminal subtype and good prognosis. J. Clin. Pathol. 2008, 61, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Pallottini, V.; Marino, M. Molecular mechanisms of action of BPA. Dose-Response 2015, 13, 1559325815610582. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Obiorah, I.; Maximov, P.Y.; Curpan, R.; Jordan, V.C. Molecular mechanism of action of bisphenol and bisphenol A mediated by oestrogen receptor alpha in growth and apoptosis of breast cancer cells. Br. J. Pharmacol. 2013, 169, 167–178. [Google Scholar] [CrossRef]
- Jalal, N.; Surendranath, A.R.; Pathak, J.L.; Yu, S.; Chung, C.Y. Bisphenol A (BPA) the mighty and the mutagenic. Toxicol. Rep. 2017, 5, 76–84. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, H.; Liu, S. Low-dose bisphenol A exposure: A seemingly instigating carcinogenic effect on breast cancer. Adv. Sci. 2016, 4, 1600248. [Google Scholar] [CrossRef]
- Papa, V.; Gliozzo, B.; Clark, G.M.; McGuire, W.L.; Moore, D.; Fujita-Yamaguchi, Y.; Vigneri, R.; Goldfine, I.D.; Pezzino, V. Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res. 1993, 53, 3736–3740. [Google Scholar]
- Resnik, J.L.; Reichart, D.B.; Huey, K.; Webster, N.J.; Seely, B.L. Elevated insulin-like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 1998, 58, 1159–1164. [Google Scholar] [PubMed]
- Chan, J.Y.; Hackel, B.J.; Yee, D. Targeting insulin receptor in breast cancer using small engineered protein scaffolds. Mol. Cancer Ther. 2017, 16, 1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to trastuzumab in breast cancer. Clin. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zi, X.; Pollak, M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int. J. Cancer 2004, 108, 334–341. [Google Scholar] [CrossRef]
- Nahta, R.; Yuan, L.X.; Zhang, B.; Kobayashi, R.; Esteva, F.J. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005, 65, 11118–11128. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, G.; Sciacca, L.; Papa, V.; Goldfine, I.D.; Vigneri, R. ASPB10 insulin induction of increased mitogenic responses and phenotypic changes in human breast epithelial cells: Evidence for enhanced interactions with the insulin-like growth factor-I receptor. Mol. Carcinog. 1997, 18, 19–25. [Google Scholar] [CrossRef]
- Drejer, K. The bioactivity of insulin analogues from in vitro receptor binding to in vivo glucose uptake. Diabetes/Metab. Rev. 1992, 8, 259–285. [Google Scholar] [CrossRef]
- Svendsen, A.M.; Winge, S.B.; Zimmermann, M.; Lindvig, A.B.; Warzecha, C.B.; Sajid, W.; Horne, M.C.; De Meyts, P. Down-regulation of cyclin G2 by insulin, IGF-I (insulin-like growth factor 1) and X10 (AspB10 insulin): Role in mitogenesis. Biochem. J. 2013, 457, 69–77. [Google Scholar] [CrossRef]
- Hvid, H.; Blouin, M.J.; Birman, E.; Damgaard, J.; Poulsen, F.; Fels, J.J.; Fledelius, C.; Hansen, B.F.; Pollak, M. Treatment with insulin analog X10 and IGF-1 increases growth of colon cancer allografts. PLoS ONE 2013, 8, e79710. [Google Scholar] [CrossRef] [Green Version]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.G.; Bland, J.M. Measurement in medicine: The analysis of method comparison studies. J. R. Stat. Society. Ser. D Stat. 1983, 32, 307–317. [Google Scholar] [CrossRef]
- Koo, T.K.; Li, M.Y. A guideline of selecting and reporting intraclass correlation coefficients for reliability research. J. Chiropr. Med. 2016, 15, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Houghton-Gisby, J.; Kerslake, R.; Karteris, E.; Mokbel, K.; Harvey, A.J. ACBD3 Bioinformatic Analysis and Protein Expression in Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8881. https://doi.org/10.3390/ijms23168881
Houghton-Gisby J, Kerslake R, Karteris E, Mokbel K, Harvey AJ. ACBD3 Bioinformatic Analysis and Protein Expression in Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(16):8881. https://doi.org/10.3390/ijms23168881
Chicago/Turabian StyleHoughton-Gisby, Jack, Rachel Kerslake, Emmanouil Karteris, Kefah Mokbel, and Amanda J. Harvey. 2022. "ACBD3 Bioinformatic Analysis and Protein Expression in Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 16: 8881. https://doi.org/10.3390/ijms23168881
APA StyleHoughton-Gisby, J., Kerslake, R., Karteris, E., Mokbel, K., & Harvey, A. J. (2022). ACBD3 Bioinformatic Analysis and Protein Expression in Breast Cancer Cells. International Journal of Molecular Sciences, 23(16), 8881. https://doi.org/10.3390/ijms23168881