
Multi-Omic Investigations of a 17–19 Translocation Links MINK1 Disruption to Autism, Epilepsy and Osteoporosis

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
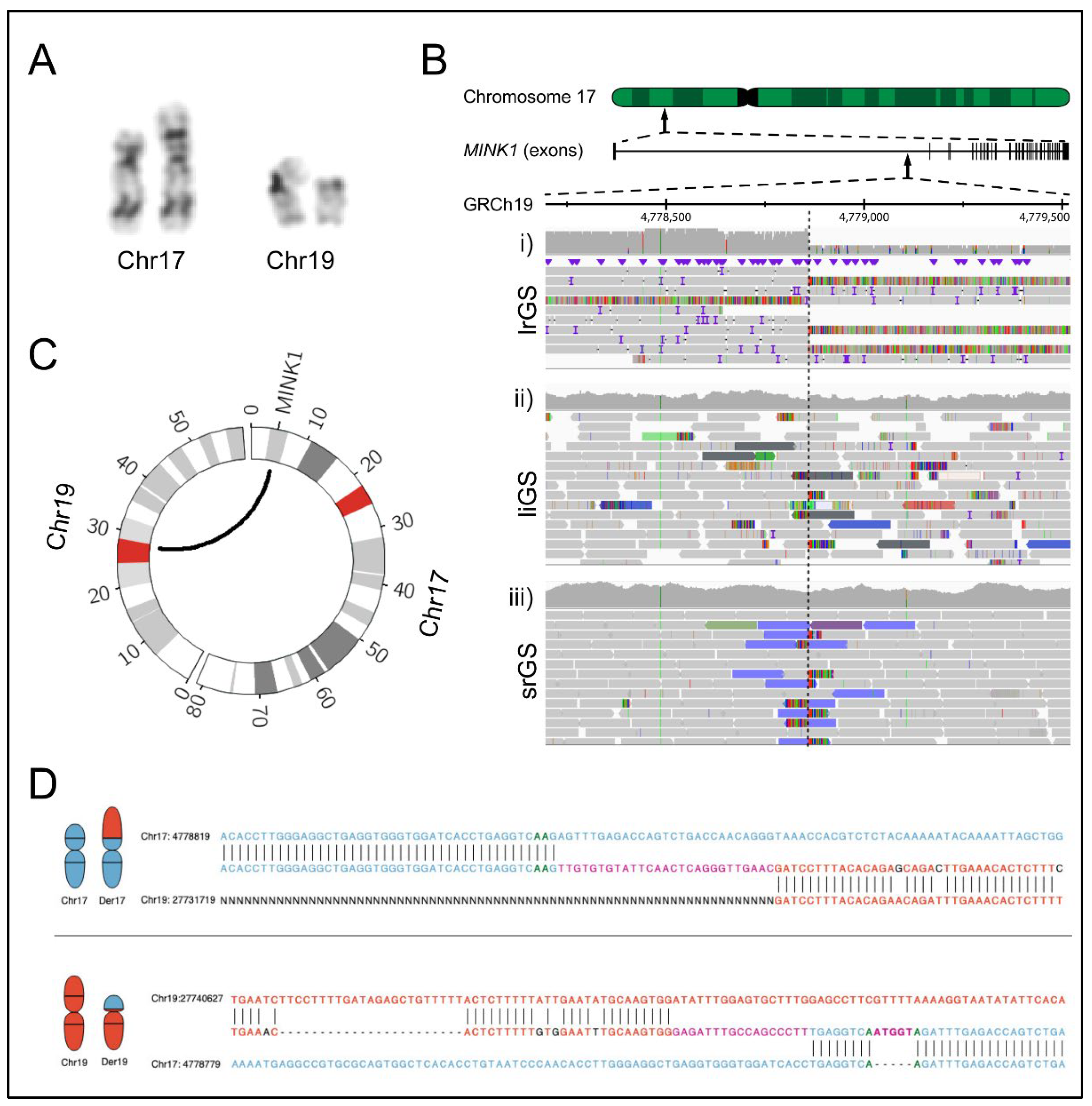
2.1. Clinical Findings
2.2. Genomic Analyses
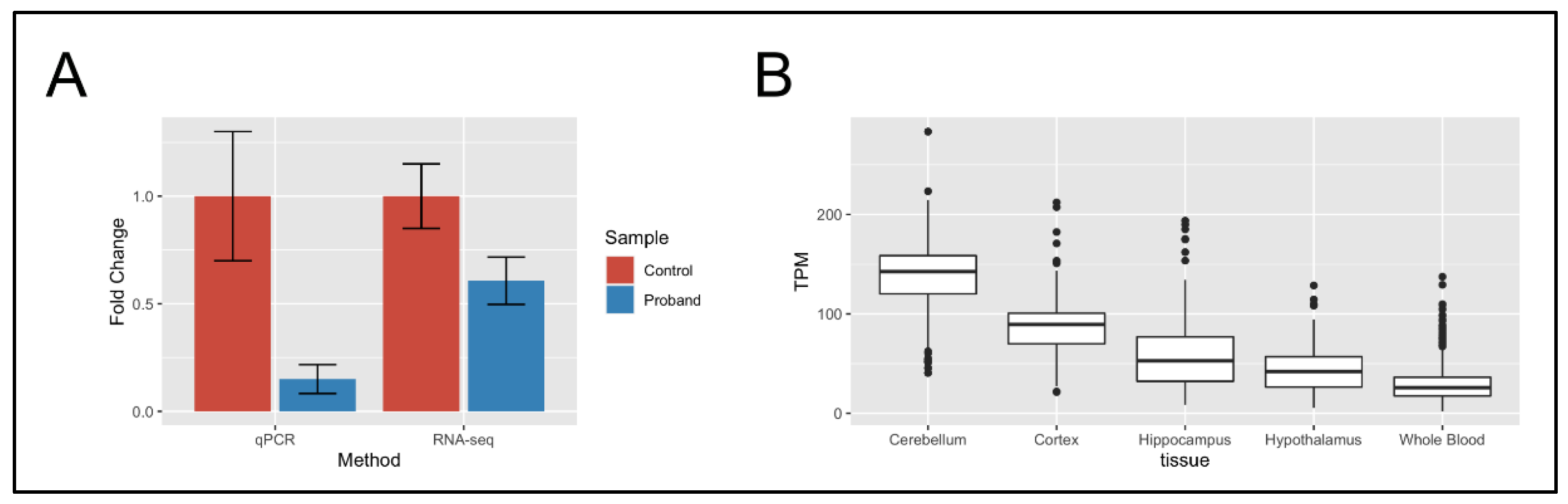
2.3. RNA-Seq of Neural Stem Cells
3. Discussion
4. Materials and Methods
4.1. Genome Analysis
4.2. Neural Stem Cell Cultivation and Transcriptome Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benko, S.; Fantes, J.A.; Amiel, J.; Kleinjan, D.J.; Thomas, S.; Ramsay, J.; Jamshidi, N.; Essafi, A.; Heaney, S.; Gordon, C.T.; et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009, 41, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, D.; Pettersson, M.; Gustavsson, P.; Forster, A.; Hofmeister, W.; Wincent, J.; Zachariadis, V.; Anderlid, B.M.; Nordgren, A.; Makitie, O.; et al. Whole-Genome Sequencing of Cytogenetically Balanced Chromosome Translocations Identifies Potentially Pathological Gene Disruptions and Highlights the Importance of Microhomology in the Mechanism of Formation. Hum. Mutat. 2017, 38, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Redin, C.; Brand, H.; Collins, R.L.; Kammin, T.; Mitchell, E.; Hodge, J.C.; Hanscom, C.; Pillalamarri, V.; Seabra, C.M.; Abbott, M.A.; et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017, 49, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Freudenberg, J. Mappability and read length. Front. Genet. 2014, 5, 381. [Google Scholar] [CrossRef] [PubMed]
- Elyanow, R.; Wu, H.T.; Raphael, B.J. Identifying structural variants using linked-read sequencing data. Bioinformatics 2018, 34, 353–360. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef]
- Li, L.; Leung, A.K.; Kwok, T.P.; Lai, Y.Y.Y.; Pang, I.K.; Chung, G.T.; Mak, A.C.Y.; Poon, A.; Chu, C.; Li, M.; et al. OMSV enables accurate and comprehensive identification of large structural variations from nanochannel-based single-molecule optical maps. Genome Biol. 2017, 18, 230. [Google Scholar] [CrossRef]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural Variation Detection and Analysis using Bionano Optical Mapping. Methods Mol. Biol. 2018, 1833, 193–203. [Google Scholar] [PubMed]
- Eisfeldt, J.; Pettersson, M.; Vezzi, F.; Wincent, J.; Kaller, M.; Gruselius, J.; Nilsson, D.; Syk Lundberg, E.; Carvalho, C.M.B.; Lindstrand, A. Comprehensive structural variation genome map of individuals carrying complex chromosomal rearrangements. PLoS Genet. 2019, 15, e1007858. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Keane, T.M.; Wong, K.; Adams, D.J. RetroSeq: Transposable element discovery from next-generation sequencing data. Bioinformatics 2013, 29, 389–390. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Sevim Bayrak, C.; Zhang, P.; Tristani-Firouzi, M.; Gelb, B.D.; Itan, Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med. 2020, 12, 9. [Google Scholar] [CrossRef]
- Larhammar, M.; Huntwork-Rodriguez, S.; Rudhard, Y.; Sengupta-Ghosh, A.; Lewcock, J.W. The Ste20 Family Kinases MAP4K4, MINK1, and TNIK Converge to Regulate Stress-Induced JNK Signaling in Neurons. J. Neurosci. 2017, 37, 11074–11084. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Hu, J.; Sheng, Z.; Fu, G.; Wang, Y.; Chen, Y.; Pan, Z.; Zhang, X.; Wu, Y.; Sun, H.; et al. Dual roles of misshapen/NIK-related kinase (MINK1) in osteoarthritis subtypes through the activation of TGFbeta signaling. Osteoarthr. Cartil. 2020, 28, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Qin, F.; Yuan, H.; He, B.; Yang, N.; Zhang, Y.; Ren, H.; Zeng, Y. Normal tissue adjacent to tumor expression profile analysis developed and validated a prognostic model based on Hippo-related genes in hepatocellular carcinoma. Cancer Med. 2021, 10, 3139–3152. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Hyodo, T.; Asano, E.; Funasaka, K.; Miyahara, R.; Hirooka, Y.; Goto, H.; Hamaguchi, M.; Senga, T. Silencing of STRN4 suppresses the malignant characteristics of cancer cells. Cancer Sci. 2014, 105, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Luo, D.; Yu, S.; Liu, P.; Zhou, Q.; Hu, M.; Liu, Y.; Wang, S.; Huang, Q.; Niu, Y.; et al. Misshapen/NIK-related kinase (MINK1) is involved in platelet function, hemostasis, and thrombus formation. Blood 2016, 127, 927–937. [Google Scholar] [CrossRef]
- Broce, I.J.; Tan, C.H.; Fan, C.C.; Jansen, I.; Savage, J.E.; Witoelar, A.; Wen, N.; Hess, C.P.; Dillon, W.P.; Glastonbury, C.M.; et al. Dissecting the genetic relationship between cardiovascular risk factors and Alzheimer’s disease. Acta Neuropathol. 2019, 137, 209–226. [Google Scholar] [CrossRef]
- Lawingco, T.; Chaudhury, S.; Brookes, K.J.; Guetta-Baranes, T.; Guerreiro, R.; Bras, J.; Hardy, J.; Francis, P.; Thomas, A.; Belbin, O.; et al. Genetic variants in glutamate-, Abeta-, and tau-related pathways determine polygenic risk for Alzheimer’s disease. Neurobiol. Aging 2021, 101, 299.e13–299.e21. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, H.; Li, X. LncRNA SNHG14 contributes to proinflammatory cytokine production in rheumatoid arthritis via the regulation of the miR-17-5p/MINK1-JNK pathway. Environ. Toxicol. 2021, 36, 2484–2492. [Google Scholar] [CrossRef]
- Kasperaviciute, D.; Catarino, C.B.; Heinzen, E.L.; Depondt, C.; Cavalleri, G.L.; Caboclo, L.O.; Tate, S.K.; Jamnadas-Khoda, J.; Chinthapalli, K.; Clayton, L.M.; et al. Common genetic variation and susceptibility to partial epilepsies: A genome-wide association study. Brain 2010, 133, 2136–2147. [Google Scholar] [CrossRef]
- Beck, T.; Shorter, T.; Brookes, A.J. GWAS Central: A comprehensive resource for the discovery and comparison of genotype and phenotype data from genome-wide association studies. Nucleic Acids Res. 2020, 48, D933–D940. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisfeldt, J.; Schuy, J.; Stattin, E.-L.; Kvarnung, M.; Falk, A.; Feuk, L.; Lindstrand, A. Multi-Omic Investigations of a 17–19 Translocation Links MINK1 Disruption to Autism, Epilepsy and Osteoporosis. Int. J. Mol. Sci. 2022, 23, 9392. https://doi.org/10.3390/ijms23169392
Eisfeldt J, Schuy J, Stattin E-L, Kvarnung M, Falk A, Feuk L, Lindstrand A. Multi-Omic Investigations of a 17–19 Translocation Links MINK1 Disruption to Autism, Epilepsy and Osteoporosis. International Journal of Molecular Sciences. 2022; 23(16):9392. https://doi.org/10.3390/ijms23169392
Chicago/Turabian StyleEisfeldt, Jesper, Jakob Schuy, Eva-Lena Stattin, Malin Kvarnung, Anna Falk, Lars Feuk, and Anna Lindstrand. 2022. "Multi-Omic Investigations of a 17–19 Translocation Links MINK1 Disruption to Autism, Epilepsy and Osteoporosis" International Journal of Molecular Sciences 23, no. 16: 9392. https://doi.org/10.3390/ijms23169392
APA StyleEisfeldt, J., Schuy, J., Stattin, E. -L., Kvarnung, M., Falk, A., Feuk, L., & Lindstrand, A. (2022). Multi-Omic Investigations of a 17–19 Translocation Links MINK1 Disruption to Autism, Epilepsy and Osteoporosis. International Journal of Molecular Sciences, 23(16), 9392. https://doi.org/10.3390/ijms23169392