
Viruses Hijack ERAD to Regulate Their Replication and Propagation

Abstract
:1. Introduction
2. The Occurrence and Development of ERAD
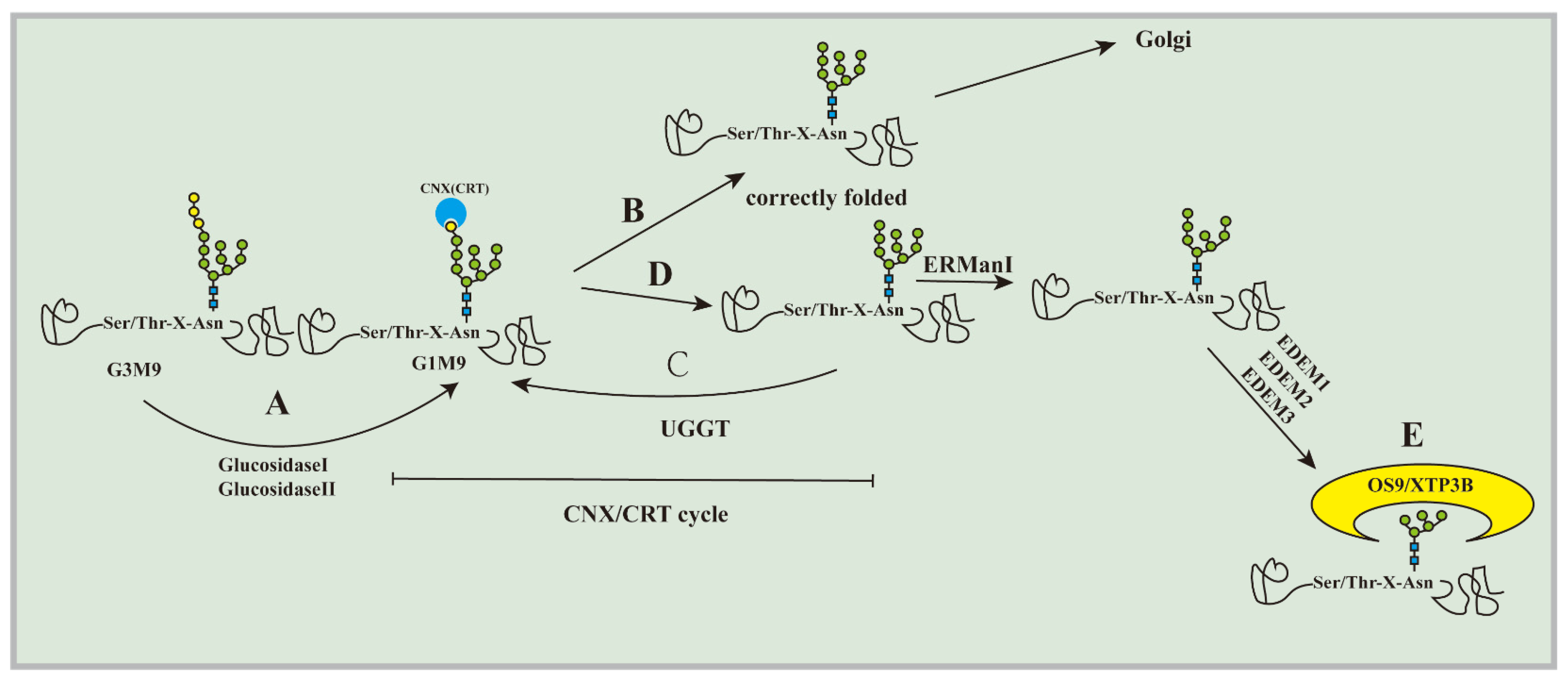
2.1. Substrate Recognition
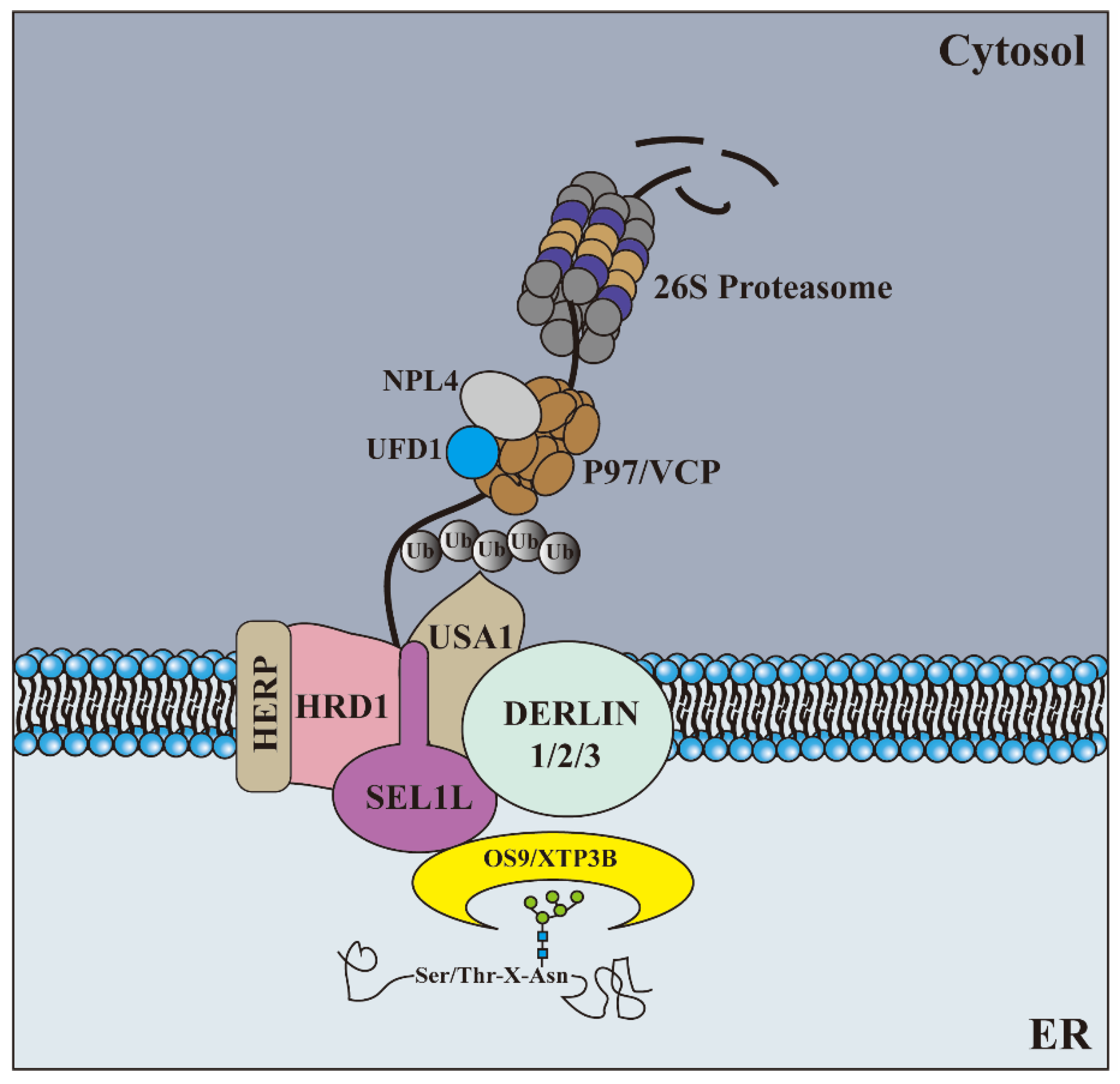
2.2. Retrotranslocation and Ubiquitination
2.3. Degradation
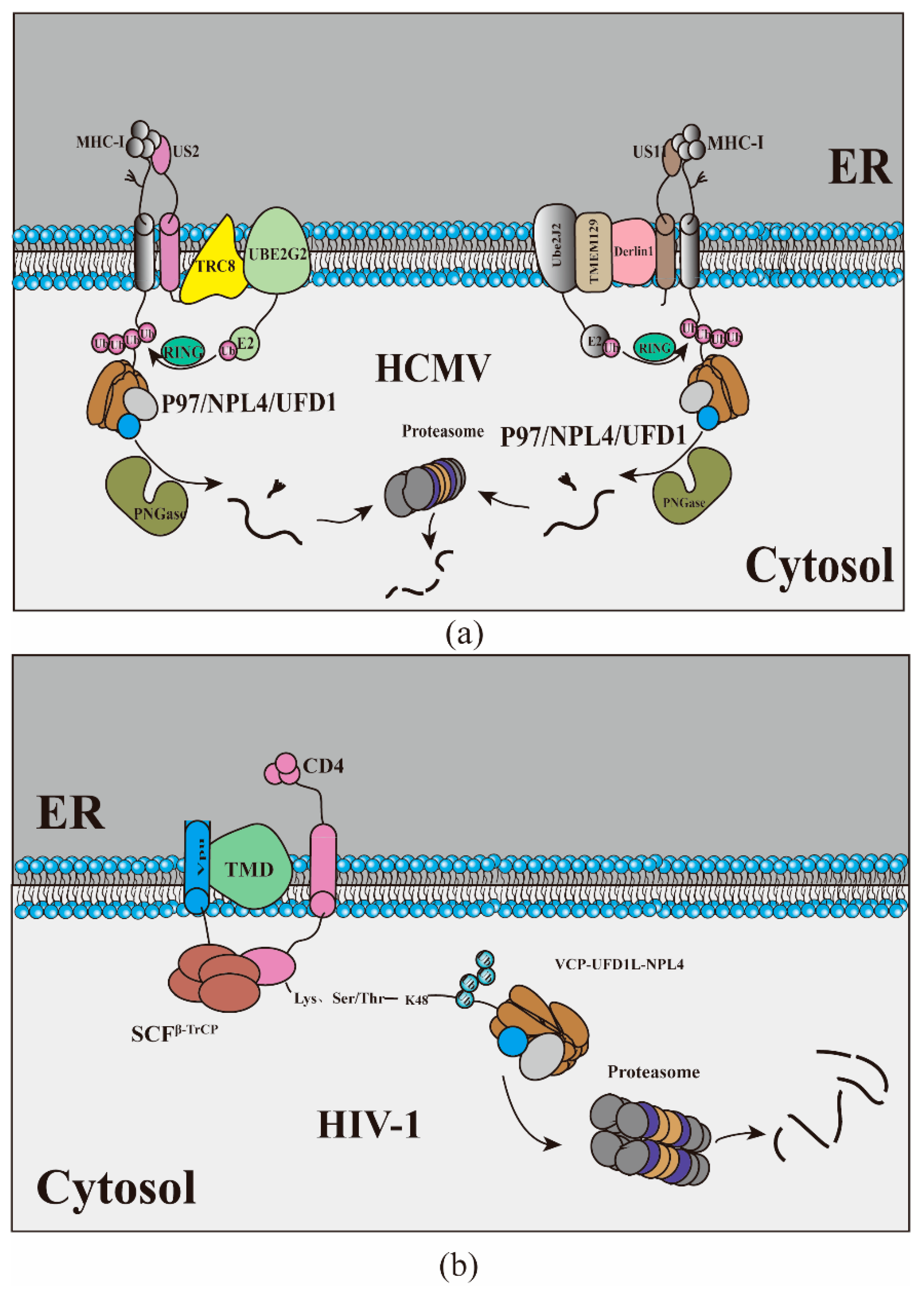
3. Viruses Hijack ERAD to Manipulate the Host Immune Response
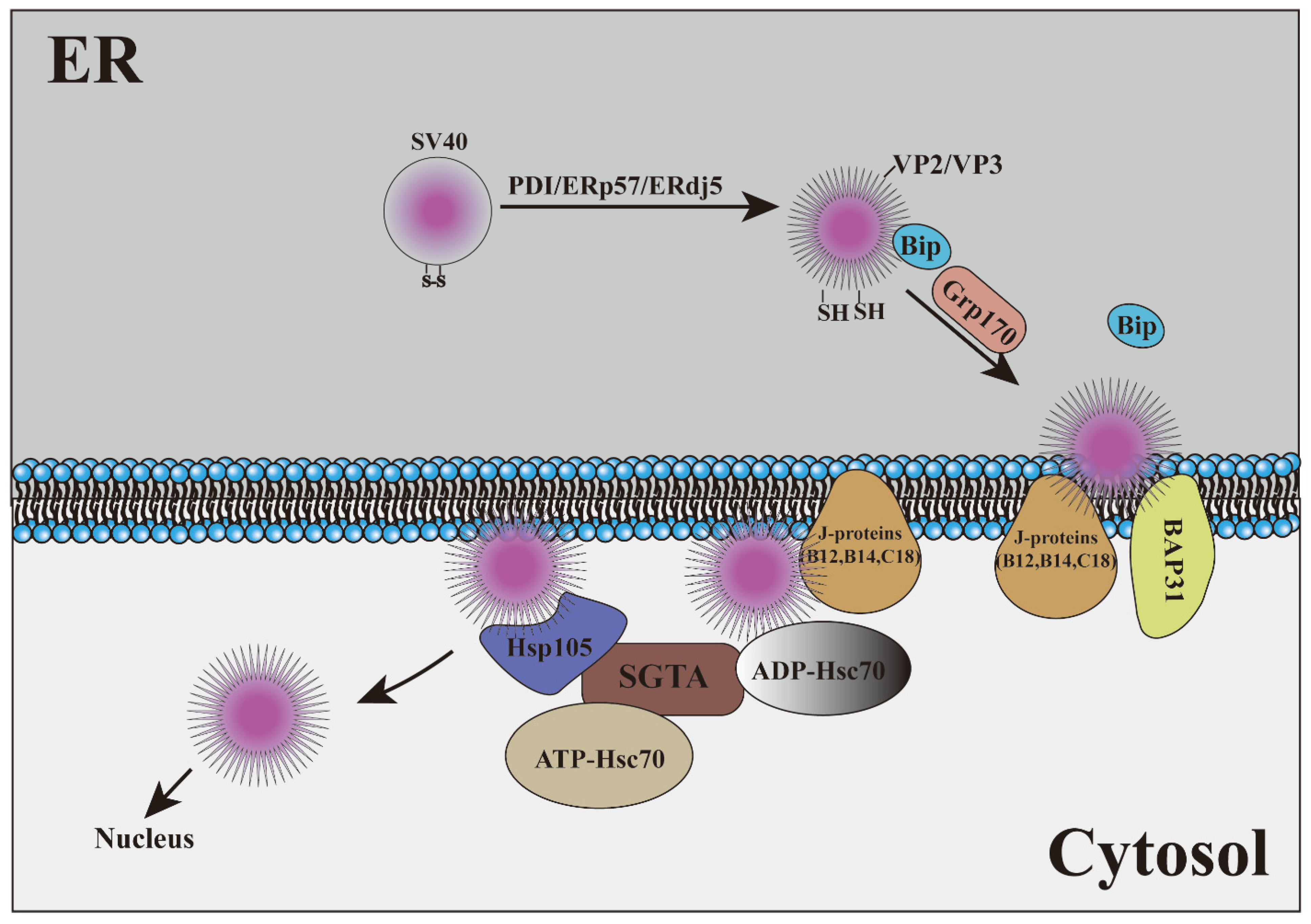
4. Viruses Hijack ERAD as a Transport Mechanism
5. Viruses Hijack ERAD to Regulate Viral Protein Expression
6. Viruses Utilize EDEMosomes as an Enclosed Safe Scaffold for Their Replication
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.L.; Mesgarzadeh, J.S.; Hendershot, L.M. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol. Cell 2022, 82, 1477–1491. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.M.; Jeon, Y.J. Proteostasis In the Endoplasmic Reticulum: Road to Cure. Cancers (Basel) 2019, 11, 1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, M.K.; Strisovsky, K. Maintenance of organellar protein homeostasis by ER-associated degradation and related mechanisms. Mol. Cell 2021, 81, 2507–2519. [Google Scholar] [CrossRef]
- Mccracken, A.A.; Brodsky, J.L. Recognition and delivery of ERAD substrates to the proteasome and alternative paths for cell survival. Curr. Top. Microbiol. Immunol. 2005, 300, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef]
- Yagishita, N.; Ohneda, K.; Amano, T.; Yamasaki, S.; Sugiura, A.; Tsuchimochi, K.; Shin, H.; Kawahara, K.; Ohneda, O.; Ohta, T.; et al. Essential role of synoviolin in embryogenesis. J. Biol. Chem. 2005, 280, 7909–7916. [Google Scholar] [CrossRef] [Green Version]
- Francisco, A.B.; Singh, R.; Li, S.; Vani, A.K.; Yang, L.; Munroe, R.J.; Diaferia, G.; Cardano, M.; Biunno, I.; Qi, L.; et al. Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J. Biol. Chem. 2010, 285, 13694–13703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eura, Y.; Yanamoto, H.; Arai, Y.; Okuda, T.; Miyata, T.; Kokame, K. Derlin-1 deficiency is embryonic lethal, Derlin-3 deficiency appears normal, and Herp deficiency is intolerant to glucose load and ischemia in mice. PLoS ONE 2012, 7, e34298. [Google Scholar] [CrossRef] [PubMed]
- Morito, D.; Nagata, K. Pathogenic Hijacking of ER-Associated Degradation: Is ERAD Flexible? Mol. Cell 2015, 59, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupzyk, A.; Tsai, B. How Polyomaviruses Exploit the ERAD Machinery to Cause Infection. Viruses 2016, 8, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Lamriben, L.; Graham, J.B.; Adams, B.M.; Hebert, D.N. N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic 2016, 17, 308–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramelo, J.J.; Castro, O.A.; Alonso, L.G.; De Prat-Gay, G.; Parodi, A.J. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc. Natl. Acad. Sci. USA 2003, 100, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.C.; Thibault, P.; Tessier, D.C.; Bergeron, J.J.; Thomas, D.Y. Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep. 2003, 4, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Caramelo, J.J.; Parodi, A.J. Getting in and out from calnexin/calreticulin cycles. J. Biol. Chem. 2008, 283, 10221–10225. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.S.; Karaveg, K.; Vandersall-Nairn, A.S.; Lal, A.; Moremen, K.W. Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J. Biol. Chem. 1999, 274, 21375–21386. [Google Scholar] [CrossRef] [Green Version]
- Olivari, S.; Cali, T.; Salo, K.E.; Paganetti, P.; Ruddock, L.W.; Molinari, M. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 2006, 349, 1278–1284. [Google Scholar] [CrossRef]
- Hosokawa, N.; Tremblay, L.O.; Sleno, B.; Kamiya, Y.; Wada, I.; Nagata, K.; Kato, K.; Herscovics, A. EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology 2010, 20, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A.; et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; You, Z.; Tremblay, L.O.; Nagata, K.; Herscovics, A. Stimulation of ERAD of misfolded null Hong Kong alpha1-antitrypsin by Golgi alpha1,2-mannosidases. Biochem. Biophys. Res. Commun. 2007, 362, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, A.; Denic, V.; Quan, E.M.; Weissman, J.S. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol. Cell 2005, 19, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, R.; Pertel, T.; Luban, J.; Molinari, M. A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: Inhibiting secretion of misfolded protein conformers and enhancing their disposal. J. Biol. Chem. 2008, 283, 16446–16454. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Wada, I.; Nagasawa, K.; Moriyama, T.; Okawa, K.; Nagata, K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008, 283, 20914–20924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, X.J.; Wang, Y.J.; Han, D.Y.; Fu, Y.L.; Duerfeldt, A.S.; Blagg, B.S.; Mu, T.W. Grp94 Protein Delivers gamma-Aminobutyric Acid Type A (GABAA) Receptors to Hrd1 Protein-mediated Endoplasmic Reticulum-associated Degradation. J. Biol. Chem. 2016, 291, 9526–9539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, J.F.; Ferro-Novick, S.; Rose, M.D.; Helenius, A. BiP/Kar2p serves as a molecular chaperone during carboxypeptidase Y folding in yeast. J. Cell Biol. 1995, 130, 41–49. [Google Scholar] [CrossRef]
- Li, Z.; Hartl, F.U.; Bracher, A. Structure and function of Hip, an attenuator of the Hsp70 chaperone cycle. Nat. Struct. Mol. Biol. 2013, 20, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Kojima, R.; Okumura, M.; Hagiwara, M.; Masui, S.; Maegawa, K.; Saiki, M.; Horibe, T.; Suzuki, M.; Inaba, K. Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding. Sci. Rep. 2013, 3, 2456. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Noi, K.; Kanemura, S.; Kinoshita, M.; Saio, T.; Inoue, Y.; Hikima, T.; Akiyama, S.; Ogura, T.; Inaba, K. Dynamic assembly of protein disulfide isomerase in catalysis of oxidative folding. Nat. Chem. Biol. 2019, 15, 499–509. [Google Scholar] [CrossRef]
- Okuda-Shimizu, Y.; Hendershot, L.M. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell 2007, 28, 544–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushioda, R.; Hoseki, J.; Nagata, K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 2013, 24, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Bridges, J.P.; Apsley, K.; Xu, Y.; Weaver, T.E. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell 2008, 19, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Tsai, B. The Grp170 nucleotide exchange factor executes a key role during ERAD of cellular misfolded clients. Mol. Biol. Cell 2016, 27, 1650–1662. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.N.; He, K.; Arunagiri, A.; Paton, A.W.; Paton, J.C.; Arvan, P.; Tsai, B. Chaperone-Driven Degradation of a Misfolded Proinsulin Mutant in Parallel With Restoration of Wild-Type Insulin Secretion. Diabetes 2017, 66, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Taxis, C.; Hitt, R.; Park, S.H.; Deak, P.M.; Kostova, Z.; Wolf, D.H. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J. Biol. Chem. 2003, 278, 35903–35913. [Google Scholar] [CrossRef] [Green Version]
- Vabulas, R.M.; Hartl, F.U. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 2005, 310, 1960–1963. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Pilon, M.; Schekman, R.; Romisch, K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997, 16, 4540–4548. [Google Scholar] [CrossRef] [Green Version]
- Romisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nakatsukasa, K.; Wigge, S.; Takano, Y.; Kawarasaki, T.; Kamura, T.; Brodsky, J.L. A positive genetic selection for transmembrane domain mutations in HRD1 underscores the importance of Hrd1 complex integrity during ERAD. Curr. Genet. 2022, 68, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.G.; Swarbrick, G.M.; Bays, N.W.; Cronin, S.R.; Wilhovsky, S.; Seelig, L.; Kim, C.; Hampton, R.Y. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 2000, 151, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Shi, G.; Han, X.; Francisco, A.B.; Ji, Y.; Mendonca, N.; Liu, X.; Locasale, J.W.; Simpson, K.W.; Duhamel, G.E.; et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 2014, 111, E582–E591. [Google Scholar] [CrossRef] [Green Version]
- Cormier, J.H.; Tamura, T.; Sunryd, J.C.; Hebert, D.N. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 2009, 34, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Saeed, M.; Suzuki, R.; Watanabe, N.; Masaki, T.; Tomonaga, M.; Muhammad, A.; Kato, T.; Matsuura, Y.; Watanabe, H.; Wakita, T.; et al. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 2011, 286, 37264–37273. [Google Scholar] [CrossRef] [Green Version]
- Gauss, R.; Jarosch, E.; Sommer, T.; Hirsch, C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat. Cell Biol. 2006, 8, 849–854. [Google Scholar] [CrossRef]
- Mueller, B.; Klemm, E.J.; Spooner, E.; Claessen, J.H.; Ploegh, H.L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12325–12330. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Olzmann, J.A.; Shaler, T.A.; Sowa, M.E.; Bennett, E.J.; Richter, C.M.; Tyler, R.E.; Greenblatt, E.J.; Harper, J.W.; Kopito, R.R. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2011, 14, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Alberts, S.M.; Sonntag, C.; Schafer, A.; Wolf, D.H. Ubx4 modulates cdc48 activity and influences degradation of misfolded proteins of the endoplasmic reticulum. J. Biol. Chem. 2009, 284, 16082–16089. [Google Scholar] [CrossRef] [Green Version]
- Vasic, V.; Denkert, N.; Schmidt, C.C.; Riedel, D.; Stein, A.; Meinecke, M. Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. 2020, 22, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Bordallo, J.; Deak, P.M.; Taxis, C.; Hitt, R.; Wolf, D.H. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 1999, 112, 4123–4134. [Google Scholar] [CrossRef]
- Mueller, B.; Lilley, B.N.; Ploegh, H.L. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 2006, 175, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, C.; Gauss, R.; Horn, S.C.; Neuber, O.; Sommer, T. The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009, 458, 453–460. [Google Scholar] [CrossRef]
- Zattas, D.; Hochstrasser, M. Ubiquitin-dependent protein degradation at the yeast endoplasmic reticulum and nuclear envelope. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Siggel, M.; Ovchinnikov, S.; Mi, W.; Svetlov, V.; Nudler, E.; Liao, M.; Hummer, G.; Rapoport, T.A. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 2020, 368, eaaz2449. [Google Scholar] [CrossRef]
- Deng, M.; Hochstrasser, M. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature 2006, 443, 827–831. [Google Scholar] [CrossRef]
- Schmidt, C.C.; Vasic, V.; Stein, A. Doa10 is a membrane protein retrotranslocase in ER-associated protein degradation. eLife 2020, 9, e56945. [Google Scholar] [CrossRef] [PubMed]
- Neal, S.; Jaeger, P.A.; Duttke, S.H.; Benner, C.; Glass, C.K.; Ideker, T.; Hampton, R.Y. The Dfm1 Derlin Is Required for ERAD Retrotranslocation of Integral Membrane Proteins. Mol. Cell 2018, 69, 306–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richly, H.; Rape, M.; Braun, S.; Rumpf, S.; Hoege, C.; Jentsch, S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 2005, 120, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Bays, N.W.; Wilhovsky, S.K.; Goradia, A.; Hodgkiss-Harlow, K.; Hampton, R.Y. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol. Biol. Cell 2001, 12, 4114–4128. [Google Scholar] [CrossRef] [Green Version]
- Rabinovich, E.; Kerem, A.; Frohlich, K.U.; Diamant, N.; Bar-Nun, S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell Biol. 2002, 22, 626–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, N.O.; Rapoport, T.A. Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 2017, 169, 722–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, 365, eaax1033. [Google Scholar] [CrossRef] [PubMed]
- Cooney, I.; Han, H.; Stewart, M.G.; Carson, R.H.; Hansen, D.T.; Iwasa, J.H.; Price, J.C.; Hill, C.P.; Shen, P.S. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 2019, 365, 502–505. [Google Scholar] [CrossRef]
- Olszewski, M.M.; Williams, C.; Dong, K.C.; Martin, A. The Cdc48 unfoldase prepares well-folded protein substrates for degradation by the 26S proteasome. Commun. Biol. 2019, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H. p97 complexes as signal integration hubs. BMC Biol. 2012, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Magadan, J.G.; Perez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010, 6, e1000869. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Ye, Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat. Struct. Mol. Biol. 2014, 21, 325–335. [Google Scholar] [CrossRef]
- Matsumura, Y.; David, L.L.; Skach, W.R. Role of Hsc70 binding cycle in CFTR folding and endoplasmic reticulum-associated degradation. Mol. Biol. Cell 2011, 22, 2797–2809. [Google Scholar] [CrossRef]
- Claessen, J.H.; Sanyal, S.; Ploegh, H.L. The chaperone BAG6 captures dislocated glycoproteins in the cytosol. PLoS ONE 2014, 9, e90204. [Google Scholar] [CrossRef] [Green Version]
- Hebert, D.N.; Simons, J.F.; Peterson, J.R.; Helenius, A. Calnexin, calreticulin, and Bip/Kar2p in protein folding. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 405–415. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, F.J.; Kikkert, M.; Wiertz, E. The HCMV gene products US2 and US11 target MHC class I molecules for degradation in the cytosol. Curr. Top. Microbiol. Immunol. 2002, 269, 37–55. [Google Scholar] [CrossRef]
- Lilley, B.N.; Ploegh, H.L. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004, 429, 834–840. [Google Scholar] [CrossRef]
- van de Weijer, M.L.; Luteijn, R.D.; Wiertz, E.J. Viral immune evasion: Lessons in MHC class I antigen presentation. Semin. Immunol. 2015, 27, 125–137. [Google Scholar] [CrossRef]
- van den Boomen, D.J.; Timms, R.T.; Grice, G.L.; Stagg, H.R.; Skodt, K.; Dougan, G.; Nathan, J.A.; Lehner, P.J. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. USA 2014, 111, 11425–11430. [Google Scholar] [CrossRef] [Green Version]
- van de Weijer, M.L.; Bassik, M.C.; Luteijn, R.D.; Voorburg, C.M.; Lohuis, M.A.; Kremmer, E.; Hoeben, R.C.; Leproust, E.M.; Chen, S.; Hoelen, H.; et al. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat. Commun. 2014, 5, 3832. [Google Scholar] [CrossRef] [Green Version]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Stagg, H.R.; Thomas, M.; van den Boomen, D.; Wiertz, E.J.; Drabkin, H.A.; Gemmill, R.M.; Lehner, P.J. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 2009, 186, 685–692. [Google Scholar] [CrossRef] [Green Version]
- van de Weijer, M.L.; Schuren, A.; van den Boomen, D.; Mulder, A.; Claas, F.; Lehner, P.J.; Lebbink, R.J.; Wiertz, E. Multiple E2 ubiquitin-conjugating enzymes regulate human cytomegalovirus US2-mediated immunoreceptor downregulation. J. Cell Sci. 2017, 130, 2883–2892. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.L.; van den Boomen, D.J.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J.; et al. Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by HCMV US2 in cooperation with UL141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef] [Green Version]
- Schuren, A.; Boer, I.; Bouma, E.M.; Van de Weijer, M.L.; Costa, A.I.; Hubel, P.; Pichlmair, A.; Lebbink, R.J.; Wiertz, E. The UFM1 Pathway Impacts HCMV US2-Mediated Degradation of HLA Class I. Molecules 2021, 26, 287. [Google Scholar] [CrossRef]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [CrossRef] [Green Version]
- Schubert, U.; Anton, L.C.; Bacik, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar] [CrossRef] [Green Version]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Nowakowska-Golacka, J.; Sominka, H.; Sowa-Rogozinska, N.; Slominska-Wojewodzka, M. Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. Int. J. Mol. Sci. 2019, 20, 1307. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Moore, P.; Tsai, B. How viruses and toxins disassemble to enter host cells. Annu. Rev. Microbiol. 2011, 65, 287–305. [Google Scholar] [CrossRef]
- Schelhaas, M.; Malmstrom, J.; Pelkmans, L.; Haugstetter, J.; Ellgaard, L.; Grunewald, K.; Helenius, A. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 2007, 131, 516–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; Lipovsky, A.; Inoue, T.; Magaldi, T.G.; Edwards, A.P.; Van Goor, K.E.; Paton, A.W.; Paton, J.C.; Atwood, W.J.; Tsai, B.; et al. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. mBio 2011, 2, e101–e111. [Google Scholar] [CrossRef] [Green Version]
- Lilley, B.N.; Gilbert, J.M.; Ploegh, H.L.; Benjamin, T.L. Murine polyomavirus requires the endoplasmic reticulum protein Derlin-2 to initiate infection. J. Virol. 2006, 80, 8739–8744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, S.M.; Jiang, M.; Imperiale, M.J. Role of cell-type-specific endoplasmic reticulum-associated degradation in polyomavirus trafficking. J. Virol. 2013, 87, 8843–8852. [Google Scholar] [CrossRef] [Green Version]
- Walczak, C.P.; Tsai, B. A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J. Virol. 2011, 85, 2386–2396. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Dosey, A.; Herbstman, J.F.; Ravindran, M.S.; Skiniotis, G.; Tsai, B. ERdj5 Reductase Cooperates with Protein Disulfide Isomerase To Promote Simian Virus 40 Endoplasmic Reticulum Membrane Translocation. J. Virol. 2015, 89, 8897–8908. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, S.I.; Fewell, S.W.; Kato, Y.; Brodsky, J.L.; Endo, T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 2001, 153, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Daniels, R.; Rusan, N.M.; Wadsworth, P.; Hebert, D.N. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: Implications for DNA translocation out of the ER. Mol. Cell 2006, 24, 955–966. [Google Scholar] [CrossRef]
- Kuksin, D.; Norkin, L.C. Disassembly of simian virus 40 during passage through the endoplasmic reticulum and in the cytoplasm. J. Virol. 2012, 86, 1555–1562. [Google Scholar] [CrossRef] [Green Version]
- Walczak, C.P.; Ravindran, M.S.; Inoue, T.; Tsai, B. A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog. 2014, 10, e1004007. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, P.; Walczak, C.P.; Tsai, B. The endoplasmic reticulum membrane J protein C18 executes a distinct role in promoting simian virus 40 membrane penetration. J. Virol. 2015, 89, 4058–4068. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, M.S.; Bagchi, P.; Inoue, T.; Tsai, B. A Non-enveloped Virus Hijacks Host Disaggregation Machinery to Translocate across the Endoplasmic Reticulum Membrane. PLoS Pathog. 2015, 11, e1005086. [Google Scholar] [CrossRef] [Green Version]
- Polier, S.; Dragovic, Z.; Hartl, F.U.; Bracher, A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell 2008, 133, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef]
- Bracher, A.; Verghese, J. GrpE, Hsp110/Grp170, HspBP1/Sil1 and BAG domain proteins: Nucleotide exchange factors for Hsp70 molecular chaperones. Subcell Biochem. 2015, 78, 1–33. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Tabata, K.; Arakawa, M.; Ishida, K.; Kobayashi, M.; Nara, A.; Sugimoto, T.; Okada, T.; Mori, K.; Morita, E. Endoplasmic Reticulum-Associated Degradation Controls Virus Protein Homeostasis, Which Is Required for Flavivirus Propagation. J. Virol. 2021, 15, e02234-20. [Google Scholar] [CrossRef]
- Rothan, H.A.; Zhong, Y.; Sanborn, M.A.; Teoh, T.C.; Ruan, J.; Yusof, R.; Hang, J.; Henderson, M.J.; Fang, S. Small molecule grp94 inhibitors block dengue and Zika virus replication. Antivir. Res. 2019, 171, 104590. [Google Scholar] [CrossRef]
- Ruan, J.; Rothan, H.A.; Zhong, Y.; Yan, W.; Henderson, M.J.; Chen, F.; Fang, S. A small molecule inhibitor of ER-to-cytosol protein dislocation exhibits anti-dengue and anti-Zika virus activity. Sci. Rep. 2019, 9, 10901. [Google Scholar] [CrossRef] [Green Version]
- Lazar, C.; Macovei, A.; Petrescu, S.; Branza-Nichita, N. Activation of ERAD pathway by human hepatitis B virus modulates viral and subviral particle production. PLoS ONE 2012, 7, e34169. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Wu, J.; Dong, M.; Shen, Z.; Lin, Y.; Li, F.; Zhang, Y.; Mao, R.; Lu, M.; et al. Host Gene SEL1L Involved in Endoplasmic Reticulum-Associated Degradation Pathway Could Inhibit Hepatitis B Virus at RNA, DNA, and Protein Levels. Front. Microbiol. 2019, 10, 2869. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.C.; Siddiquey, M.; Zhang, H.; Li, G.; Kamil, J.P. Human Cytomegalovirus Tropism Modulator UL148 Interacts with SEL1L, a Cellular Factor That Governs Endoplasmic Reticulum-Associated Degradation of the Viral Envelope Glycoprotein gO. J. Virol. 2018, 92, e00688-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashiba, M.; Collins, D.R.; Terry, V.H.; Collins, K.L. Vpr Overcomes Macrophage-Specific Restriction of HIV-1 Env Expression and Virion Production. Cell Host Microbe 2015, 17, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhou, T.; Frabutt, D.A.; Zheng, Y.H. HIV-1 Vpr increases Env expression by preventing Env from endoplasmic reticulum-associated protein degradation (ERAD). Virology 2016, 496, 194–202. [Google Scholar] [CrossRef]
- Zhou, T.; Dang, Y.; Zheng, Y.H. The mitochondrial translocator protein, TSPO, inhibits HIV-1 envelope glycoprotein biosynthesis via the endoplasmic reticulum-associated protein degradation pathway. J. Virol. 2014, 88, 3474–3484. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Frabutt, D.A.; Moremen, K.W.; Zheng, Y.H. ERManI (Endoplasmic Reticulum Class I alpha-Mannosidase) Is Required for HIV-1 Envelope Glycoprotein Degradation via Endoplasmic Reticulum-associated Protein Degradation Pathway. J. Biol. Chem. 2015, 290, 22184–22192. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of assembly: Roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. Microbiol. 2008, 6, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Cali, T.; Galli, C.; Olivari, S.; Molinari, M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 2008, 371, 405–410. [Google Scholar] [CrossRef]
- Merulla, J.; Fasana, E.; Solda, T.; Molinari, M. Specificity and regulation of the endoplasmic reticulum-associated degradation machinery. Traffic 2013, 14, 767–777. [Google Scholar] [CrossRef]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salonen, A.; Ahola, T.; Kaariainen, L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005, 285, 139–173. [Google Scholar] [CrossRef]
- Neuman, B.W.; Angelini, M.M.; Buchmeier, M.J. Does form meet function in the coronavirus replicative organelle? Trends Microbiol. 2014, 22, 642–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, R.; Noack, J.; Molinari, M. Unconventional roles of nonlipidated LC3 in ERAD tuning and coronavirus infection. Autophagy 2012, 8, 1534–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monastyrska, I.; Ulasli, M.; Rottier, P.J.; Guan, J.L.; Reggiori, F.; de Haan, C.A. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy 2013, 9, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Bhattacharyya, S.; Nain, M.; Kaur, M.; Sood, V.; Gupta, V.; Khasa, R.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 2014, 10, 1637–1651. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component (Yeast) | Component (Mammals) | Function | References |
|---|---|---|---|
| Kar2 | Bip | Substrate recognition and recruitment | [75] |
| Cne1 | Calnexin (CNX) | Lectin chaperone | [14,15] |
| Calreticulin (CRT) | |||
| - | UGGT1 | Glycoprotein glucosyltransferase | [17] |
| UGGT2 | |||
| Mns1 | Man1B1 (ERMan Ⅰ) | N-glycan trimming from M9 | [19] |
| Htm1 | EDEM1 | N-glycan trimming fromM8 to M7 | [20,21] |
| EDEM2 | N-glycan trimming fromM9 to M8 | [23] | |
| EDEM3 | N-glycan trimming fromM8 to M7 | [22,23] | |
| Yos9 | OS-9 | Recognize a terminal α1,6-linked mannosyl residue | [24] |
| XTP3 | |||
| Hrd1 | HRD1 | Retrotranslocation channel | [44,59] |
| gp78 | |||
| Hrd3 | SEL1L | Substrate recognition and recruitment | [46] |
| Der1 | Derlin1 | Retrotranslocation channel | [59] |
| Derlin2 | |||
| Derlin3 | |||
| Doa10 | Teb-4/MARCH6 | Retrotranslocation channel | [60] |
| Cdc48 | p97/VCP | Substrates dislocation | [63] |
| Ufd1 | UFD1 | Cofactor of p97 | [52] |
| Npl4 | NPL4 | Cofactor of p97 | [52] |
| Virus | ERAD Component | Mechanism | References |
|---|---|---|---|
| HCMV | TMEM129, Derlin1, Ube2j2 | US11 recruits TMEM129, and TMEM129 recruit Ube2J2 driving MHC-I to cytoplasm, then deglycosylated by PNGase | [77,78,79,80] |
| TRC8, Ube2g2 | TRC8 bind to US2, resulting in polyubiquitin of MHC-I, | [83] | |
| HIV | VCP, UFD1 L, NPL4 | Vpu targets CD4 receptors and rapidly degrades CD4 | [86,87] |
| SV40 | PDI, ERp57, ERdj5 | SV40 VP2 binds to BAP31 to stabilize the membrane-embedded virus and then SV40 is transport to the cytoplasm under the action of ER transmembrane J-proteins | [98,101,102] |
| DENV | Derlin2, grp94, VCP | avoid excessive accumulation of nonstructural protein | [108,109,110] |
| ZIKV | HRD1 | avoid excessive accumulation of nonstructural protein | [109,110] |
| JEV | VCP | avoid excessive accumulation of nonstructural protein | [108] |
| HCV | EDEM1, EDEM2, EDEM3 | IRE1 induces ERAD to degrade nonstructural protein | [48] |
| HBV | EDEM1, SEL1L | degrade nonstructural protein | [111,112] |
| MHV | EDEMosome | nsp2 and nsp3 make RTC near to ER and induces EDEMosome, DMV, CM and DMS | [121,124] |
| EAV | EDEMosome | utilize EDEMosome as replication sites | [125] |
| SARS-CoV | EDEMosome | nsp3/4 induces DMV construct | [124,125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, L.; Wang, X.; Zhao, F.; Wu, K.; Li, X.; Li, Z.; Li, Y.; Chen, W.; Zeng, S.; Liu, X.; et al. Viruses Hijack ERAD to Regulate Their Replication and Propagation. Int. J. Mol. Sci. 2022, 23, 9398. https://doi.org/10.3390/ijms23169398
Zou L, Wang X, Zhao F, Wu K, Li X, Li Z, Li Y, Chen W, Zeng S, Liu X, et al. Viruses Hijack ERAD to Regulate Their Replication and Propagation. International Journal of Molecular Sciences. 2022; 23(16):9398. https://doi.org/10.3390/ijms23169398
Chicago/Turabian StyleZou, Linke, Xinyan Wang, Feifan Zhao, Keke Wu, Xiaowen Li, Zhaoyao Li, Yuwan Li, Wenxian Chen, Sen Zeng, Xiaodi Liu, and et al. 2022. "Viruses Hijack ERAD to Regulate Their Replication and Propagation" International Journal of Molecular Sciences 23, no. 16: 9398. https://doi.org/10.3390/ijms23169398
APA StyleZou, L., Wang, X., Zhao, F., Wu, K., Li, X., Li, Z., Li, Y., Chen, W., Zeng, S., Liu, X., Zhao, M., Yi, L., Fan, S., & Chen, J. (2022). Viruses Hijack ERAD to Regulate Their Replication and Propagation. International Journal of Molecular Sciences, 23(16), 9398. https://doi.org/10.3390/ijms23169398