
In Vivo Evaluation of Praziquantel-Loaded Solid Lipid Nanoparticles against S. mansoni Infection in Preclinical Murine Models





Abstract
:1. Introduction
2. Results
2.1. Physical Analysis of the Nanoparticulate System
2.2. Evaluation of FTIR Spectroscopy
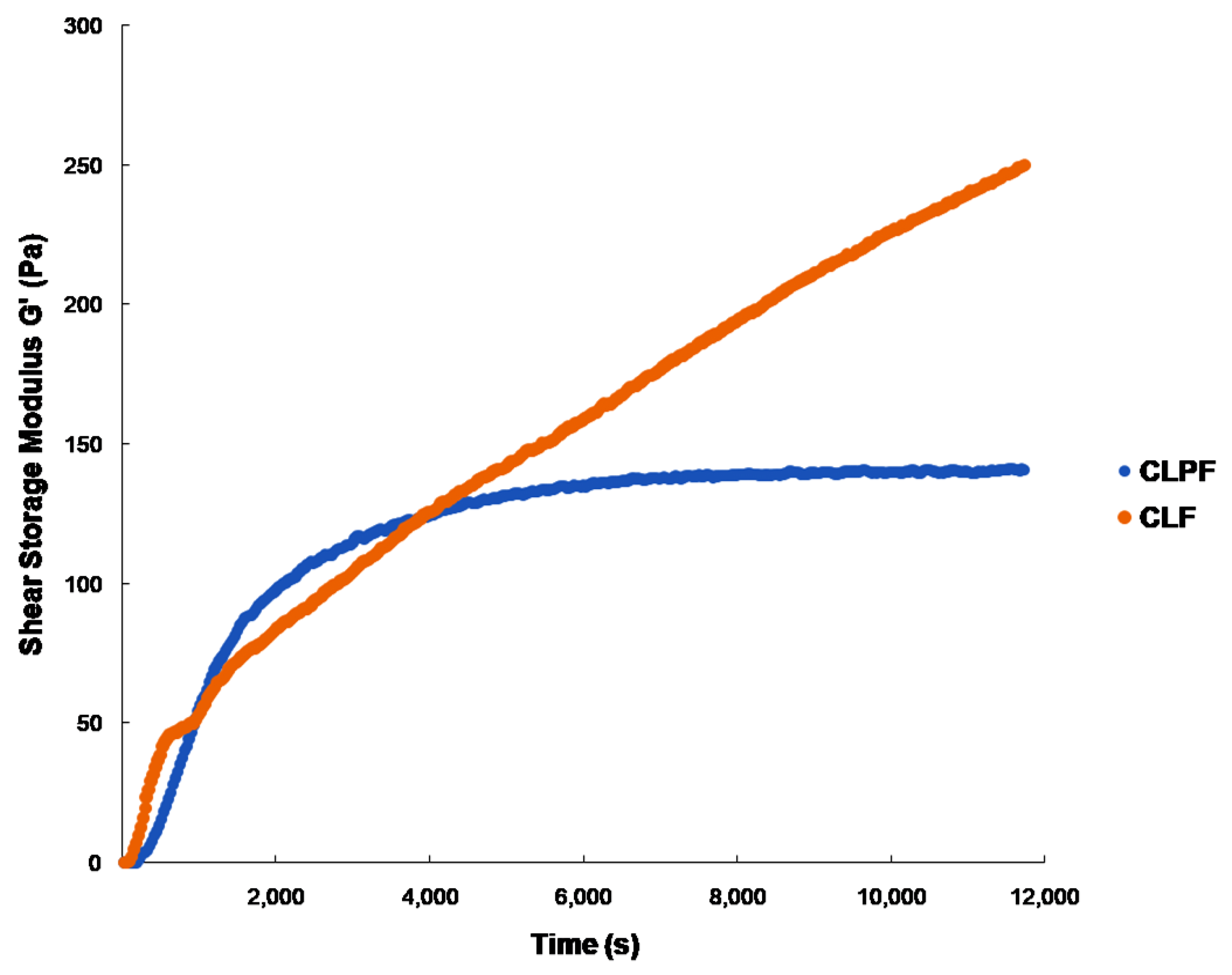
2.3. Evaluation of Mechanical Properties under Physiological Conditions
2.4. Evaluation of Crystal Nature of the Formulated SLNs
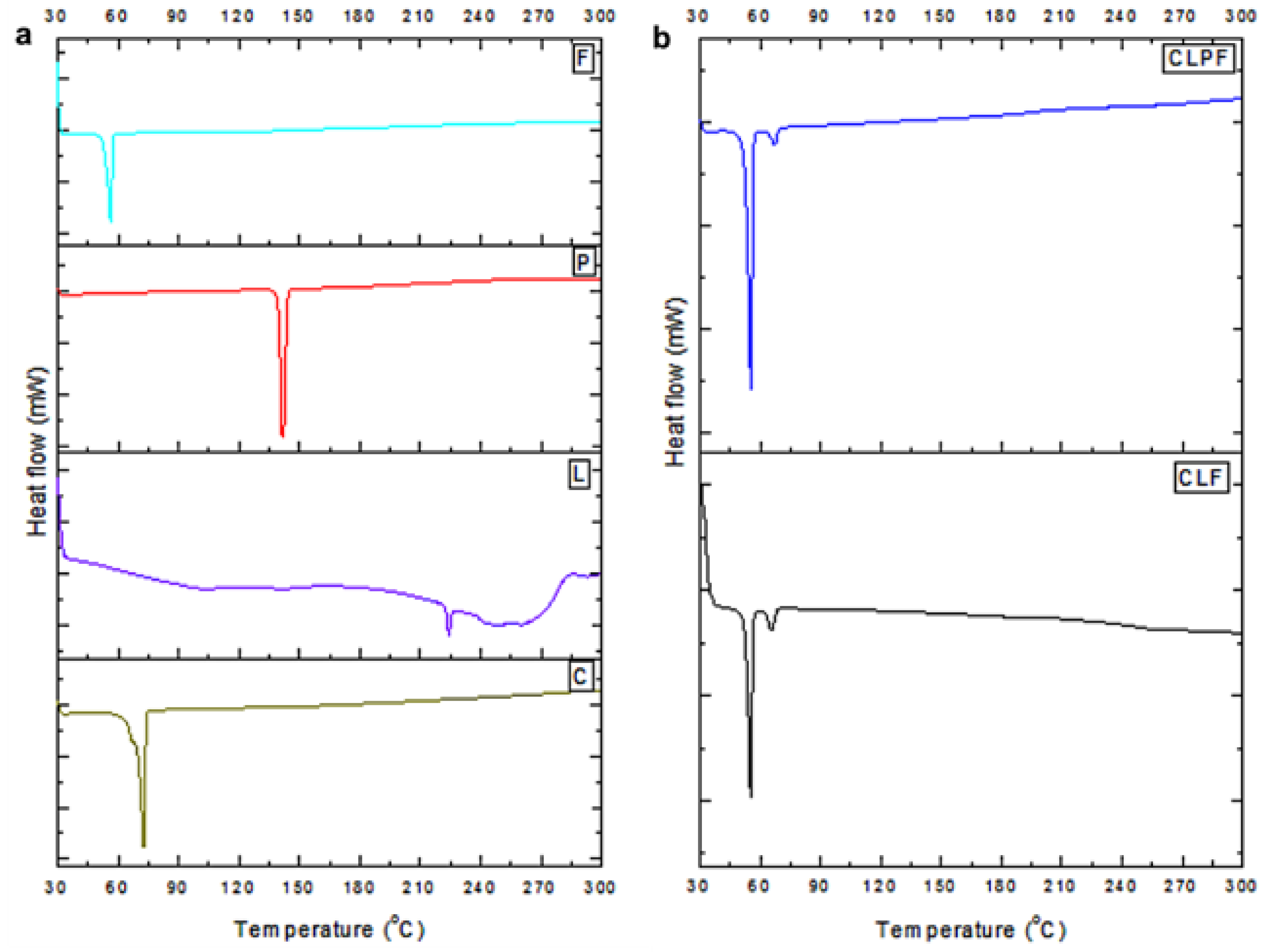
2.5. Evaluation of Thermophysical Properties of the Formulated SLNs
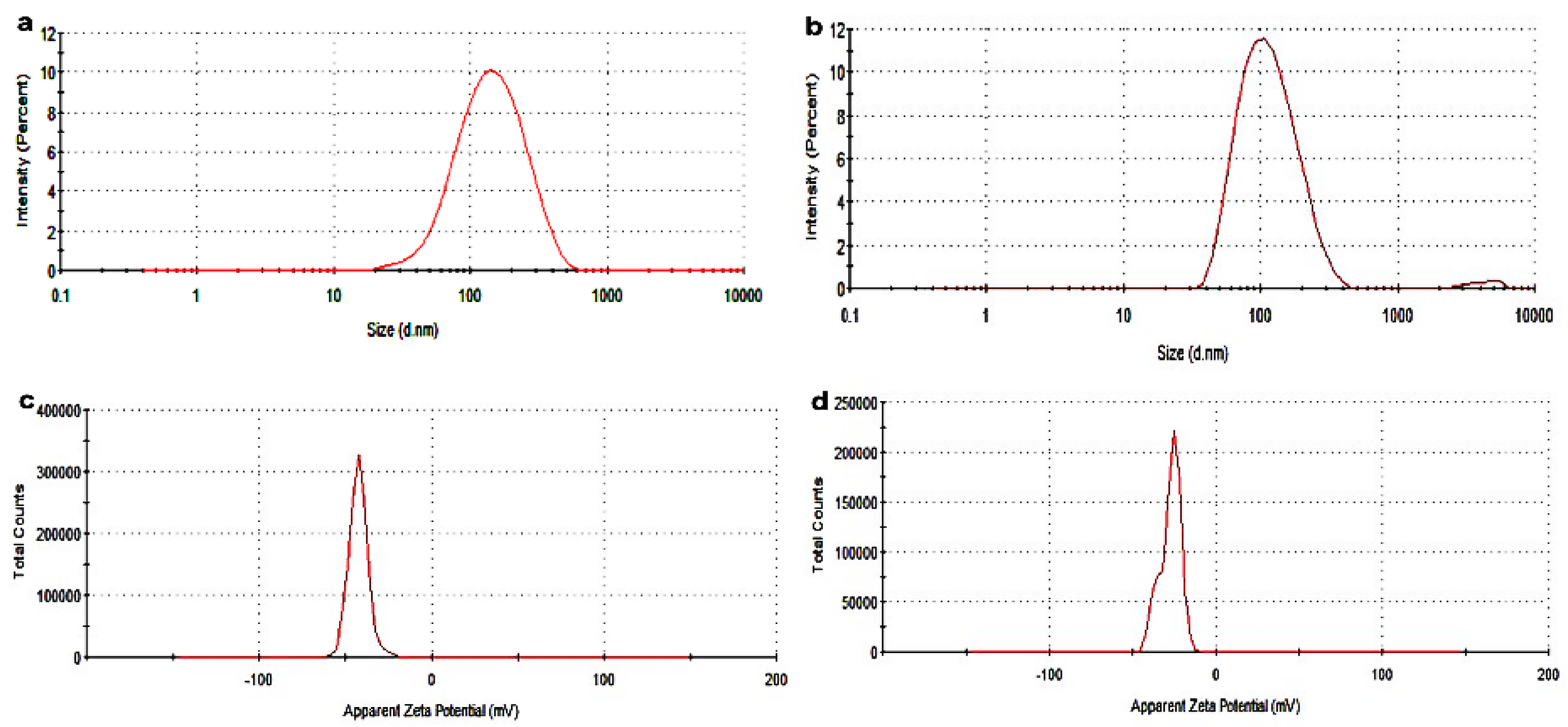
2.6. Investigation of the Colloidal System of the Synthesized SLNs
2.7. Analysis of the In Vitro Release Behaviour of PZQ from the Formulated CLPF-SLN
2.8. Particle Size Distribution, PDI, and Zeta Potential Analysis of the SLNs as a Function of Time
2.9. Stability Analysis of Unloaded and PZQ-Loaded CLF-SLN through Turbiscan Technology as a Function of Time
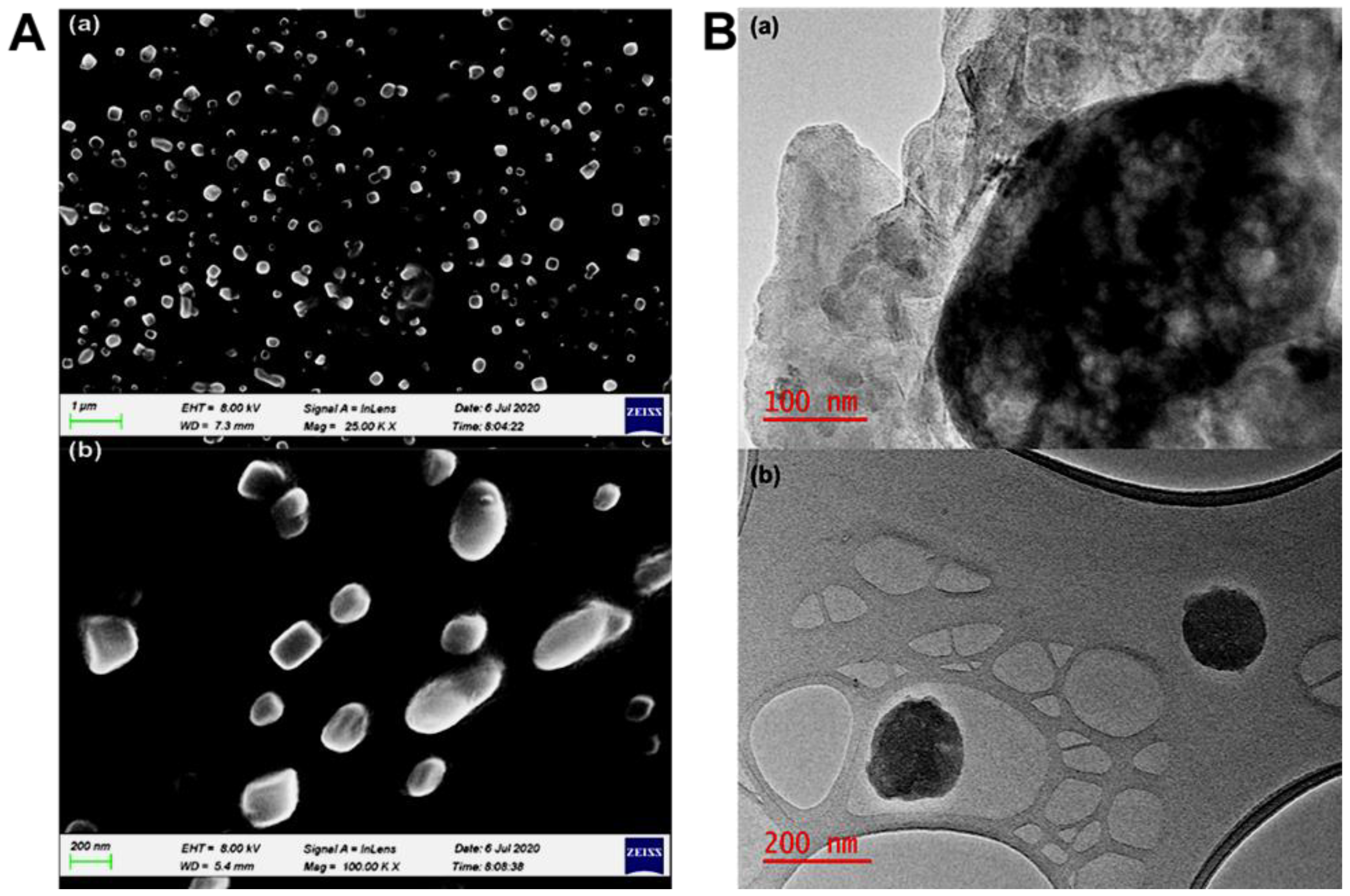
2.10. Surface Morphology Characterization of the SLNs after 120 Days
2.11. Analysis of In Vitro Cytotoxicity of SLNs
2.12. Analysis of RAW 264.7 Murine Macrophage Cells Morphology
2.13. In Vivo Toxicity Evaluation
2.14. Histopathological Analysis
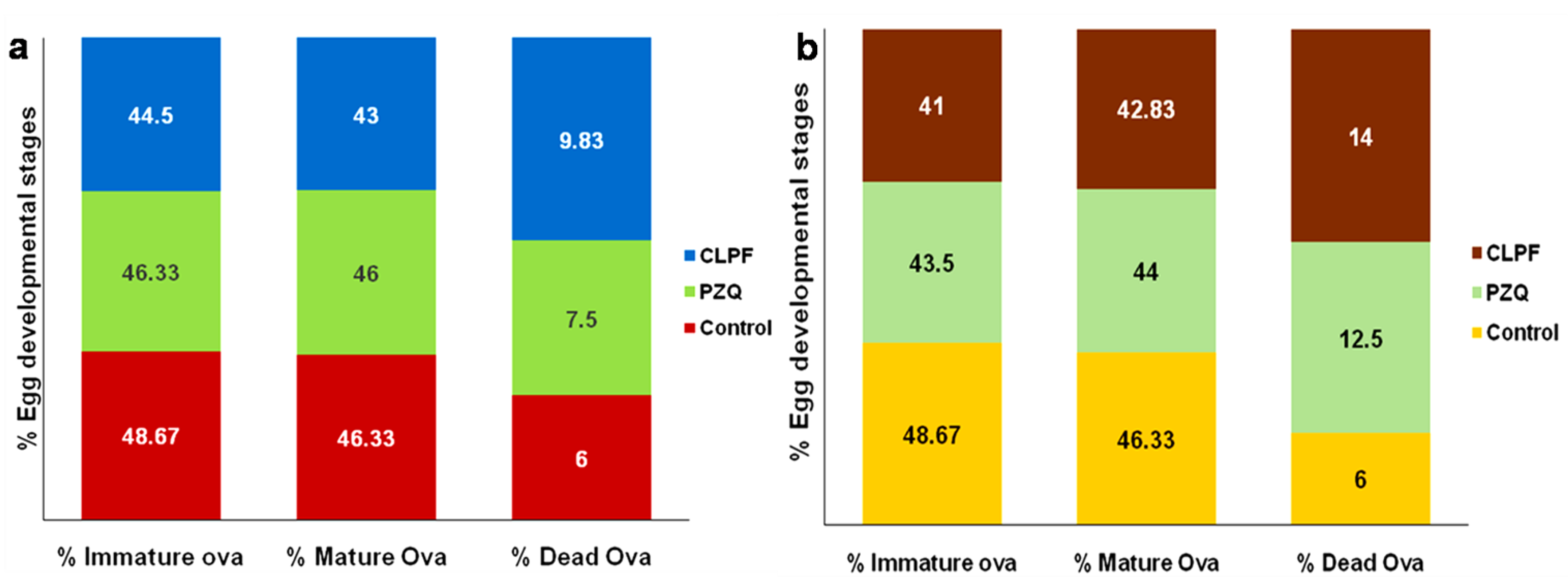
2.15. Evaluation of Parasitological Cure Rate
3. Discussion
4. Materials and Methods
4.1. Material and Reagents
4.2. Preparation of Compritol-Lecithin SLN-Loaded Praziquantel
4.3. Determination of Particle Size Distribution, Polydispersity Index (PDI), and Zeta Potential
4.4. Evaluation of the Drug Entrapment Efficacy and Drug Loading Capacity
4.5. Evaluation of Fourier Transform Infrared Spectroscopy (FTIR)
4.6. Mechanical Properties Analysis
4.7. X-ray Powder Diffraction (XRPD) Evaluation
4.8. Analysis of Differential Scanning Calorimetry (DSC)
4.9. Colloidal System Analysis of the Formulated SLNs
4.10. In Vitro Analysis of PZQ Release from the Formulated SLNs
4.11. Optical Characterization and Stability Profiling of the Suspensions of SLNs Formulation
4.12. Scanning Electron Microscopy and Transmission Electron Microscopy and
4.13. In Vitro Cytotoxicity Assay (MTT Assay)
4.14. Evaluation of Cell Morphology
4.15. In Vivo Toxicity Evaluation
4.16. In Vivo Parasitological Study
4.16.1. Infection of Animals
4.16.2. Experimental Design
- Group 1: Infected control.
- Group 2: Single dose 250 mg/kg (PZQ equivalent) CLPF was administered two weeks post-infection.
- Group 3: A single dosage of PZQ 250 mg/kg was administered two weeks post-infection.
- Group 4: A single dosage of 250 mg/kg of CLPF was administered four weeks post-infection.
- Group 5: A single dosage of PZQ 250 mg/kg was administered four weeks post-infection.
4.16.3. Assessment of Parasitological Cure Rate
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kohn, A.B.; Anderson, P.A.; Roberts-Misterly, J.M.; Greenberg, R.M. Schistosome calcium channel β subunits unusual modulatory effects and potential role in the action of the antischistosomal drug praziquantel. J. Biol. Chem. 2001, 276, 36873–36876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vale, N.; Gouveia, M.J.; Rinaldi, G.; Brindley, P.J.; Gärtner, F.; Correia da Costa, J.M. Praziquantel for schistosomiasis: Single-drug metabolism revisited, mode of action, and resistance. Antimicrob. Agents Chemother. 2017, 61, e02582-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aruleba, R.T.; Adekiya, T.A.; Oyinloye, B.E.; Masamba, P.; Mbatha, L.S.; Pretorius, A.; Kappo, A.P. PZQ therapy: How close are we in the development of effective alternative antischistosomal drugs? Infect. Disord. Drug Targets 2019, 19, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Adekiya, T.A.; Kondiah, P.P.; Choonara, Y.E.; Kumar, P.; Pillay, V. A review of nanotechnology for targeted antischistosomal therapy. Front. Bioeng. Biotechnol. 2020, 8, 32. [Google Scholar] [CrossRef]
- Mourão, S.C.; Costa, P.I.; Salgado, H.R.; Gremião, M.P.D. Improvement of antischistosomal activity of praziquantel by incorporation into phosphatidylcholine-containing liposomes. Int. J. Pharm. 2005, 295, 157–162. [Google Scholar] [CrossRef]
- Frezza, T.F.; Gremião, M.P.D.; Zanotti-Magalhães, E.M.; Magalhães, L.A.; de Souza, A.L.R.; Allegretti, S.M. Liposomal-praziquantel: Efficacy against Schistosoma mansoni in a preclinical assay. Acta Trop. 2013, 128, 70–75. [Google Scholar] [CrossRef]
- El-Feky, G.S.; Mohamed, W.S.; Nasr, H.E.; El-Lakkany, N.M.; Seif El-Din, S.H.; Botros, S.S. Praziquantel in a clay nanoformulation shows more bioavailability and higher efficacy against murine Schistosoma mansoni infection. Antimicrob. Agents Chemother. 2015, 59, 3501–3508. [Google Scholar] [CrossRef] [Green Version]
- Adekiya, T.A.; Kumar, P.; Kondiah, P.P.D.; Choonara, Y.E. In Vivo Evaluation of an Antibody-Functionalized Lipoidal Nanosystem for Schistosomiasis Intervention. Pharmaceutics 2022, 14, 1531. [Google Scholar] [CrossRef]
- Kushwaha, A.K.; Vuddanda, P.R.; Karunanidhi, P.; Singh, S.K.; Singh, S. Development and evaluation of solid lipid nanoparticles of raloxifene hydrochloride for enhanced bioavailability. BioMed Res. Int. 2013, 2013, 584549. [Google Scholar] [CrossRef] [Green Version]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Baboota, S.; Ali, J.; Khan, S.; Narang, R.S.; Narang, J.K. Nanostructured lipid carriers: An emerging platform for improving oral bioavailability of lipophilic drugs. Int. J. Pharm. Investig. 2015, 5, 182. [Google Scholar]
- Bayón-Cordero, L.; Alkorta, I.; Arana, L. Application of solid lipid nanoparticles to improve the efficiency of anticancer drugs. Nanomaterials 2019, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Garnero, C.; Chattah, A.K.; Aloisio, C.; Fabietti, L.; Longhi, M. Improving the stability and the pharmaceutical properties of norfloxacin form C through binary complexes with β-cyclodextrin. AAPS PharmSciTech 2018, 19, 2255–2263. [Google Scholar] [CrossRef]
- Chuacharoen, T.; Sabliov, C.M. Stability and controlled release of lutein loaded in zein nanoparticles with and without lecithin and pluronic F127 surfactants. Colloids Surf. A Physicochem. Eng. Asp. 2016, 503, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Shaarani, S.; Hamid, S.S.; Kaus, N.H.M. The Influence of pluronic F68 and F127 nanocarrier on physicochemical properties, in vitro release, and antiproliferative activity of thymoquinone drug. Pharmacogn. Res. 2017, 9, 12. [Google Scholar]
- Sokolsky-Papkov, M.; Kabanov, A. Synthesis of well-defined gold nanoparticles using pluronic: The role of radicals and surfactants in nanoparticles formation. Polymers 2019, 11, 1553. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.S.; Yagnesh, T.N.S. Pharmaceutical suspensions: Patient compliance oral dosage forms. World J. Pharm. Pharm. Sci. 2016, 5, 1471–1537. [Google Scholar]
- Üstündağ-Okur, N.; Yurdasiper, A.; Gündoğdu, E.; Homan Gökçe, E. Modification of solid lipid nanoparticles loaded with nebivolol hydrochloride for improvement of oral bioavailability in treatment of hypertension: Polyethylene glycol versus chitosan oligosaccharide lactate. J. Microencapsul. 2016, 33, 30–42. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, A.; Garg, N. Acoustic cavitation-assisted formulation of solid lipid nanoparticles using different stabilizers. ACS Omega 2019, 4, 13360–13370. [Google Scholar] [CrossRef] [Green Version]
- Sumaila, M.; Ramburrun, P.; Kumar, P.; Choonara, Y.E.; Pillay, V. Lipopolysaccharide polyelectrolyte complex for oral delivery of an anti-tubercular drug. AAPS PharmSciTech 2019, 20, 107. [Google Scholar] [CrossRef]
- Campos, F.D.S.; Cassimiro, D.L.; Crespi, M.S.; Almeida, A.E.; Gremião, M.P.D. Preparation and characterisation of Dextran-70 hydrogel for controlled release of praziquantel. Braz. J. Pharm. Sci. 2013, 49, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, X.; Wang, J.K.; Ching, C.B. Structural characterization and enantioseparation of the chiral compound praziquantel. J. Pharm. Sci. 2004, 93, 3039–3046. [Google Scholar] [CrossRef] [PubMed]
- Zanolla, D.; Hasa, D.; Arhangelskis, M.; Schneider-Rauber, G.; Chierotti, M.R.; Keiser, J.; Voinovich, D.; Jones, W.; Perissutti, B. Mechanochemical formation of racemic praziquantel hemihydrate with improved biopharmaceutical properties. Pharmaceutics 2020, 12, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok, A.; Kumar, A.; Tarlochan, F. Colloidal metal oxide nanocrystals in catalysis. In Colloidal Metal Oxide Nanoparticles; Elsevier: Amsterdam, The Netherlands, 2020; pp. 247–288. [Google Scholar]
- Chhabra, R.P.; Gurappa, B. (Eds.) Coulson and Richardson’s Chemical Engineering: Volume 2A: Particulate Systems and Particle Technology; Butterworth-Heinemann: Oxford, UK, 2019. [Google Scholar]
- Dora, C.P.; Kushwah, V.; Katiyar, S.S.; Kumar, P.; Pillay, V.; Suresh, S.; Jain, S. Improved metabolic stability and therapeutic efficacy of a novel molecular gemcitabine phospholipid complex. Int. J. Pharm. 2017, 530, 113–127. [Google Scholar] [CrossRef]
- Ibrahim, W.H.; Habib, H.M.; Jarrar, A.H.; Al Baz, S.A. Effect of Ramadan fasting on markers of oxidative stress and serum biochemical markers of cellular damage in healthy subjects. Ann. Nutr. Metab. 2008, 53, 175–181. [Google Scholar] [CrossRef]
- Adekiya, T.A.; Kumar, P.; Kondiah, P.P.; Pillay, V.; Choonara, Y.E. Synthesis and therapeutic delivery approaches for praziquantel: A patent review (2010–present). Expert Opin. Ther. Patents 2021, 31, 851–865. [Google Scholar] [CrossRef]
- Murthy, S.N.; Shivakumar, H.N. Topical and transdermal drug delivery. In Handbook of Non-Invasive Drug Delivery Systems; William Andrew Publishing: Norwich, NY, USA, 2010; pp. 1–36. [Google Scholar]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; HasanzadehDavarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, G.; Raut, P. Pharmaceutical Compositions. WO2015071668A1, 21 May 2015. [Google Scholar]
- Sun, Q.; Mao, R.; Wang, D.; Hu, C.; Zheng, Y.; Sun, D. The cytotoxicity study of praziquantel enantiomers. Drug Des. Dev. Ther. 2016, 10, 2061. [Google Scholar] [CrossRef] [Green Version]
- Aburahma, M.H.; Badr-Eldin, S.M. Compritol 888 ATO: A multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin. Drug Deliv. 2014, 11, 1865–1883. [Google Scholar] [CrossRef]
- Lippens, E.; Swennen, I.; Gironès, J.; Declercq, H.; Vertenten, G.; Vlaminck, L.; Gasthuys, F.; Schacht, E.; Cornelissen, R. Cell survival and proliferation after encapsulation in a chemically modified Pluronic® F127 hydrogel. J. Biomater. Appl. 2013, 27, 828–839. [Google Scholar] [CrossRef]
- Thornton, C.G.; MacLellan, K.M.; Brink, T.L., Jr.; Wolfe, D.M.; Llorin, O.J.; Passen, S. Processing respiratory specimens with C18-carboxypropylbetaine: Development of a sediment resuspension buffer that contains lytic enzymes to reduce the contamination rate and lecithin to alleviate toxicity. J. Clin. Microbiol. 1998, 36, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Tallima, H.; Salah, M.; El Ridi, R. In vitro and in vivo effects of unsaturated fatty acids on Schistosoma mansoni and S. haematobium lung-stage larvae. J. Parasitol. 2005, 91, 1094–1102. [Google Scholar] [CrossRef]
- Ceccaldi, C.; Strandman, S.; Hui, E.; Montagnon, E.; Schmitt, C.; Henni, A.H.; Lerouge, S. Validation and application of a nondestructive and contactless method for rheological evaluation of biomaterials. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 2565–2573. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.S.; Bruce, J.I.; Boyd, D.A. Laboratory cultivation of schistosome vector snails and maintenance of schistosome life cycles. In Proceedings of the 1st Sino-American Symposium held at the International Conference Hall National Yangming Medical College, Taipei, Taiwan, 30 September–4 October 1985; pp. 34–48. [Google Scholar]
- Christensen, N.; Gotsche, G.; Frandsen, F. Parasitological technique for use in laboratory maintenance of schistosomes and for use in studies on the epidemiology of human and bovine schistosomiasis. In Danish Bilharziasis Laboratory Manual; Danish Bilharziasis Laboratory: Copenhagen, Denmark, 1984; p. 112. [Google Scholar]
- Pellegrino, J.; Oliveira, C.A.; Faria, J.; Cunha, A.S. New approach to the screening of drugs in experimental schistosomiasis mansoni in mice. Am. J. Trop. Med. Hyg. 1962, 11, 201–215. [Google Scholar] [CrossRef]
- Kloetzel, K. A suggestion for the prevention of severe clinical forms of Schistosomiasis mansoni. Bull. World Health Organ. 1967, 37, 686. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Sample | Size (nm) | PDI (a.u) | Zeta Potential (mV) | %DEE | %LC |
|---|---|---|---|---|---|---|
| 1 | CLF-SLN | 101.6 ± 0.7 | 0.27 ± 0.006 | −23.6 ± 0.26 | - | - |
| 2 | CLPF-SLN | 112.9 ± 1.0 | 0.23 ± 0.010 | −19.0 ± 0.26 | 71.63 ± 0.3 | 11.46 ± 0.61 |
| S/N | Composition | T (Onset)/°C | T (Peak)/°C | ΔH (J/g) |
|---|---|---|---|---|
| 1 | PF127 (F) | 54.02 | 55.84 | −101.37 |
| 2 | PZQ (P) | 139.60 | 141.37 | −115.10 |
| 3 | Compritol (C) | 69.08 | 72.29 | −124.52 |
| 4 | Lecithin (L) | 222.85 | 223.99 | −1.95 |
| 5 | Comp-Lec-F127 (CLF) | 52.64 | 55.22 | −56.33 |
| 6 | Comp-Lec-PZQ-F127(CLPF) | 52.57 | 55.38 | −65.05 |
| Mean Worm Burden ± SD (Liver and Porto-Mesenteric) | % Reduction in Total Worm Burden | ||||
|---|---|---|---|---|---|
| Male | Female | Couples | Total | ||
| Control | 2.33 ± 0.81 | 0.33 ± 0.52 | 6.17 ± 0.75 | 15.00 ± 0.89 | |
| PZQ | 1.33 ± 1.21 | 0 | 4.33 ± 0.82 | 10.00 ± 1.41 | 33.30 |
| CLPF | 1.50 ± 0.55 | 0 | 3.67 ± 1.21 | 8.83 ± 2.64 | 41.13 |
| Mice Group | Liver | % Reduction in Ova Count in Liver | Intestine | % Reduction in Ova Count in the Intestine |
|---|---|---|---|---|
| Control | 28,202 ± 4372 | 31,902 ± 4342 | ||
| PZQ | 20,303 ± 2175 | 28.00 | 22,702 ± 5347 | 28.84 |
| CLPF | 16,548 ± 6919 | 41.32 | 21,120 ± 6644 | 33.79 |
| Mean Worm Burden ± SD (Liver and Porto-Mesenteric) | % Reduction in Total Worm Burden | ||||
|---|---|---|---|---|---|
| Male | Female | Couples | Total | ||
| Control | 2.33 ± 0.81 | 0.33 ± 0.52 | 6.17 ± 0.75 | 15.00 ± 0.89 | |
| PZQ | 1.50 ± 1.40 | 0 | 2.00 ± 0.89 | 5.50 ± 2.60 | 63.30 |
| CLPF | 1.00 ± 0.63 | 0 | 1.33 ± 1.37 | 4.33 ± 1.86 | 71.13 |
| Mice Group | Liver | % Reduction in Ova Count in Liver | Intestine | % Reduction in Ova Count in Intestine |
|---|---|---|---|---|
| Control | 28,202 ± 4372 | 31,902 ± 4342 | ||
| PZQ | 13,626 ± 2936 | 51.68 | 14,658 ± 3699 | 54.05 |
| CLPF | 11,384 ± 4135 | 59.60 | 10,310 ± 1080 | 67.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adekiya, T.A.; Kumar, P.; Kondiah, P.P.D.; Ubanako, P.; Choonara, Y.E. In Vivo Evaluation of Praziquantel-Loaded Solid Lipid Nanoparticles against S. mansoni Infection in Preclinical Murine Models. Int. J. Mol. Sci. 2022, 23, 9485. https://doi.org/10.3390/ijms23169485
Adekiya TA, Kumar P, Kondiah PPD, Ubanako P, Choonara YE. In Vivo Evaluation of Praziquantel-Loaded Solid Lipid Nanoparticles against S. mansoni Infection in Preclinical Murine Models. International Journal of Molecular Sciences. 2022; 23(16):9485. https://doi.org/10.3390/ijms23169485
Chicago/Turabian StyleAdekiya, Tayo A., Pradeep Kumar, Pierre P. D. Kondiah, Philemon Ubanako, and Yahya E. Choonara. 2022. "In Vivo Evaluation of Praziquantel-Loaded Solid Lipid Nanoparticles against S. mansoni Infection in Preclinical Murine Models" International Journal of Molecular Sciences 23, no. 16: 9485. https://doi.org/10.3390/ijms23169485
APA StyleAdekiya, T. A., Kumar, P., Kondiah, P. P. D., Ubanako, P., & Choonara, Y. E. (2022). In Vivo Evaluation of Praziquantel-Loaded Solid Lipid Nanoparticles against S. mansoni Infection in Preclinical Murine Models. International Journal of Molecular Sciences, 23(16), 9485. https://doi.org/10.3390/ijms23169485