


Interplay between the Chd4/NuRD Complex and the Transcription Factor Znf219 Controls Cardiac Cell Identity

 , ,
, ,
Abstract
:1. Introduction
2. Results
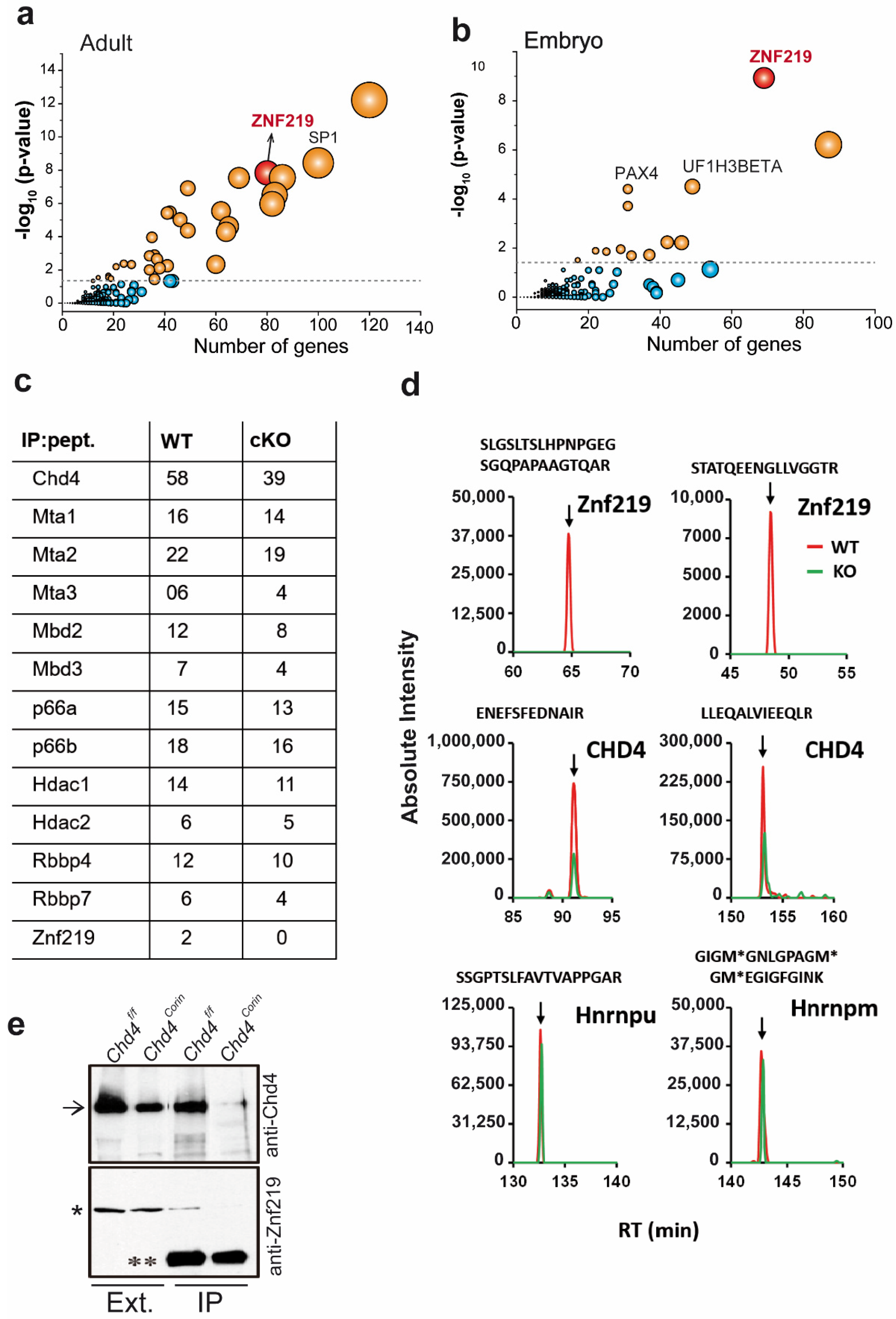
2.1. Promoter Regions of the Genes Upregulated in Chd4-Mutant Hearts Are Enriched in Znf219 DNA-Binding Sites
2.2. The Chd4/NuRD Complex and Znf219 Physically Interact in the Heart
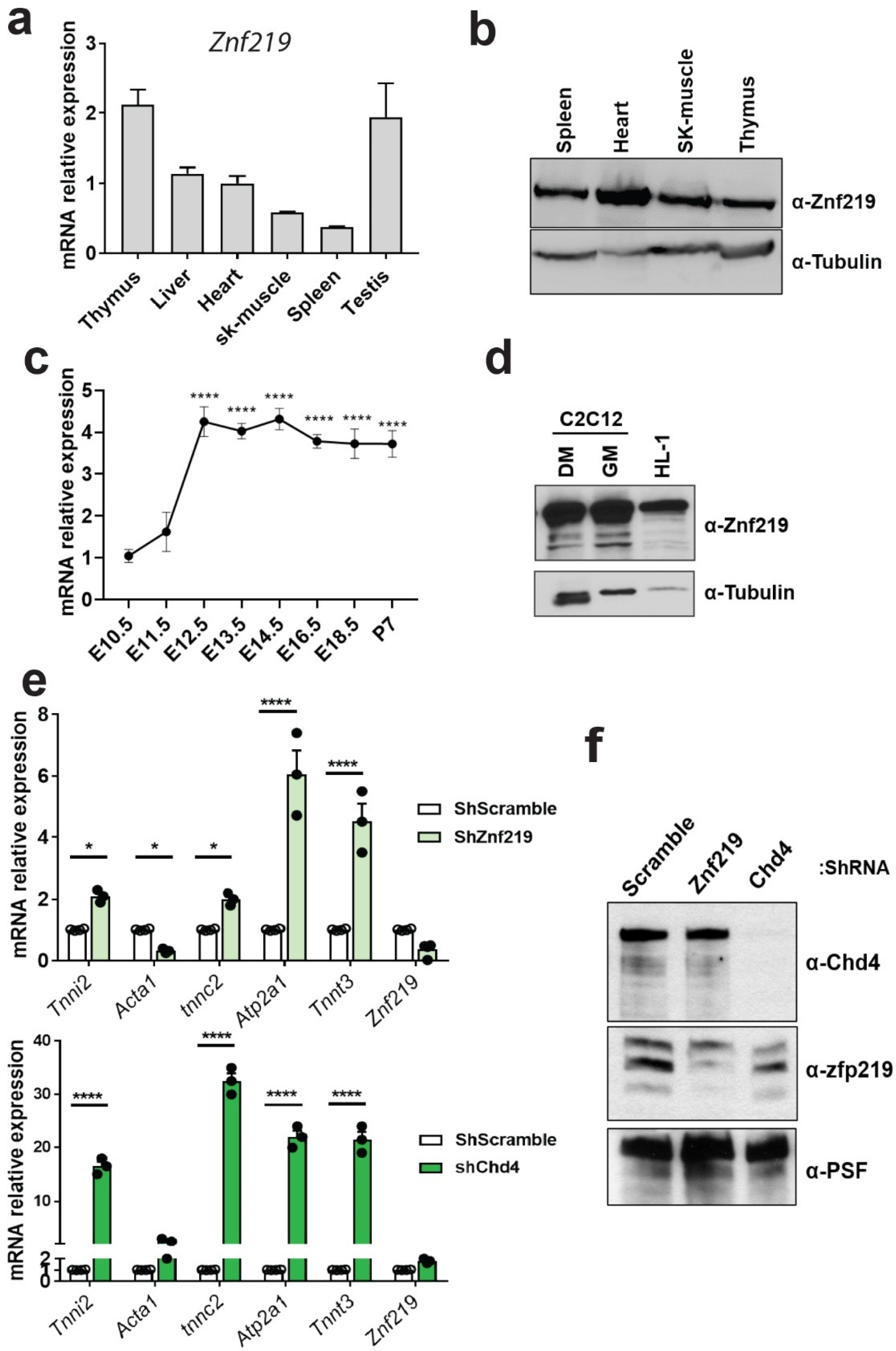
2.3. Znf219 Is Highly Expressed in Mouse Heart and in Striated Muscle Cell Lines and Is Involved in the Repression of the Skeletal-Muscle Gene Program In Vitro
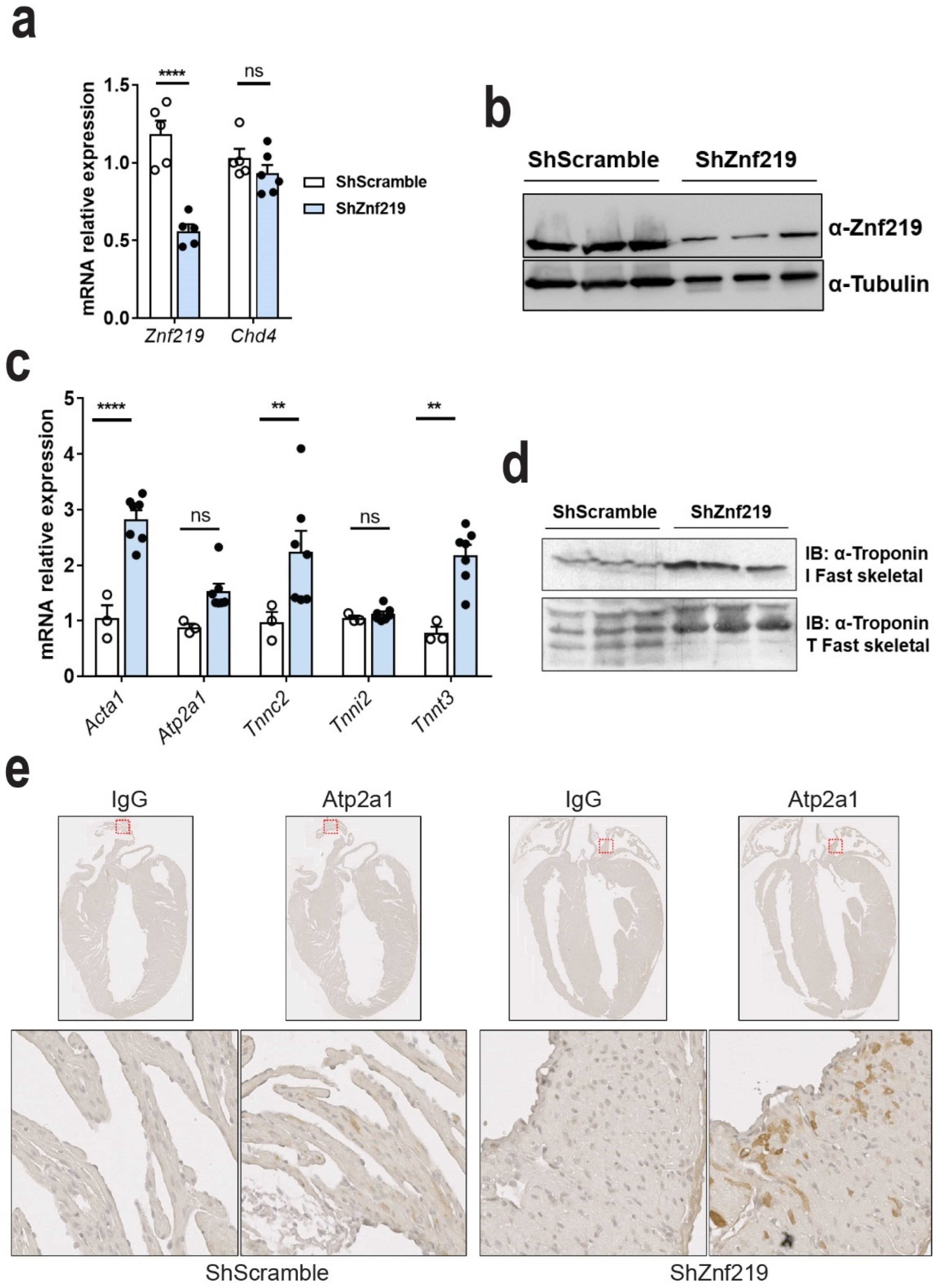
2.4. Znf219 Represses the Skeletal Muscle Gene Program in the Heart
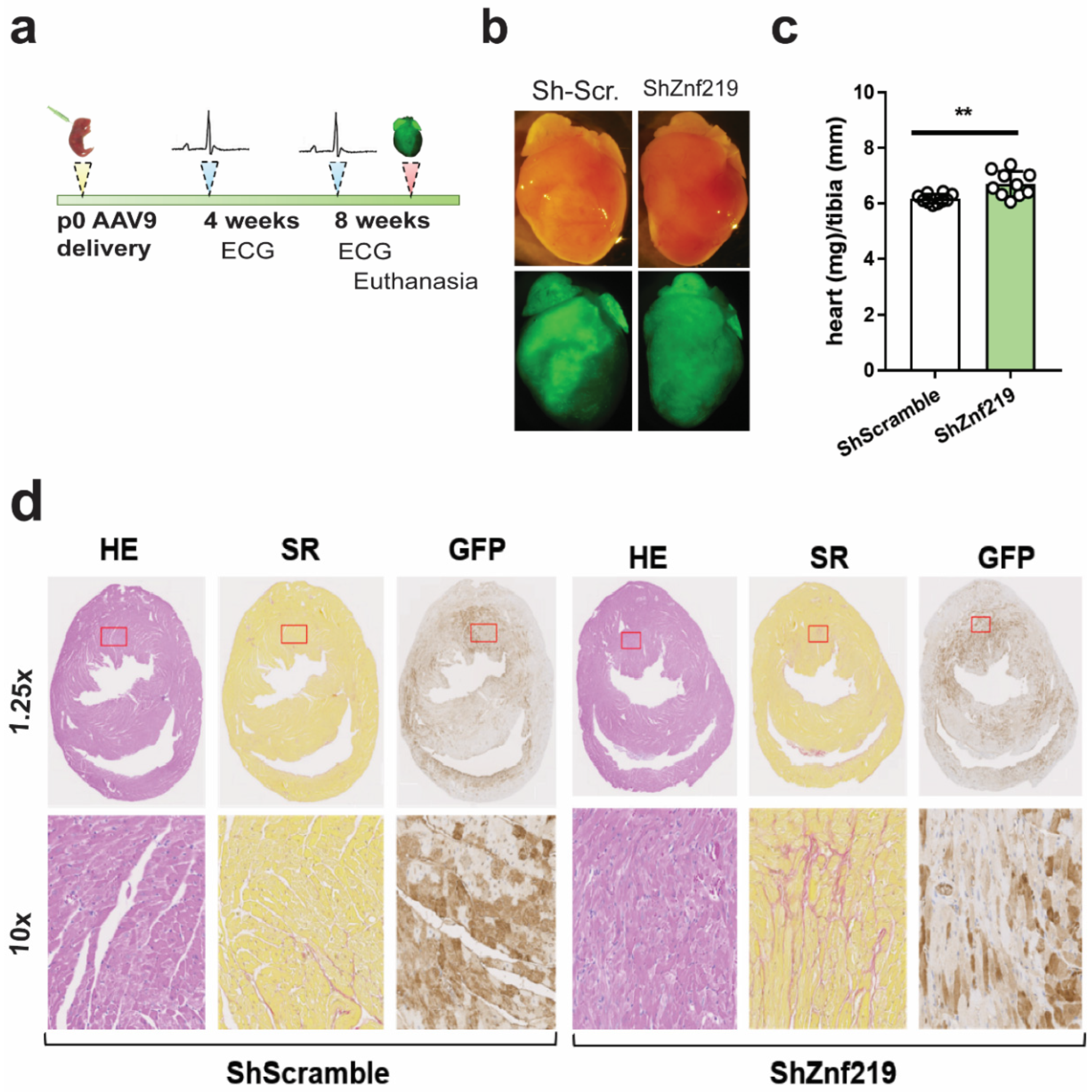
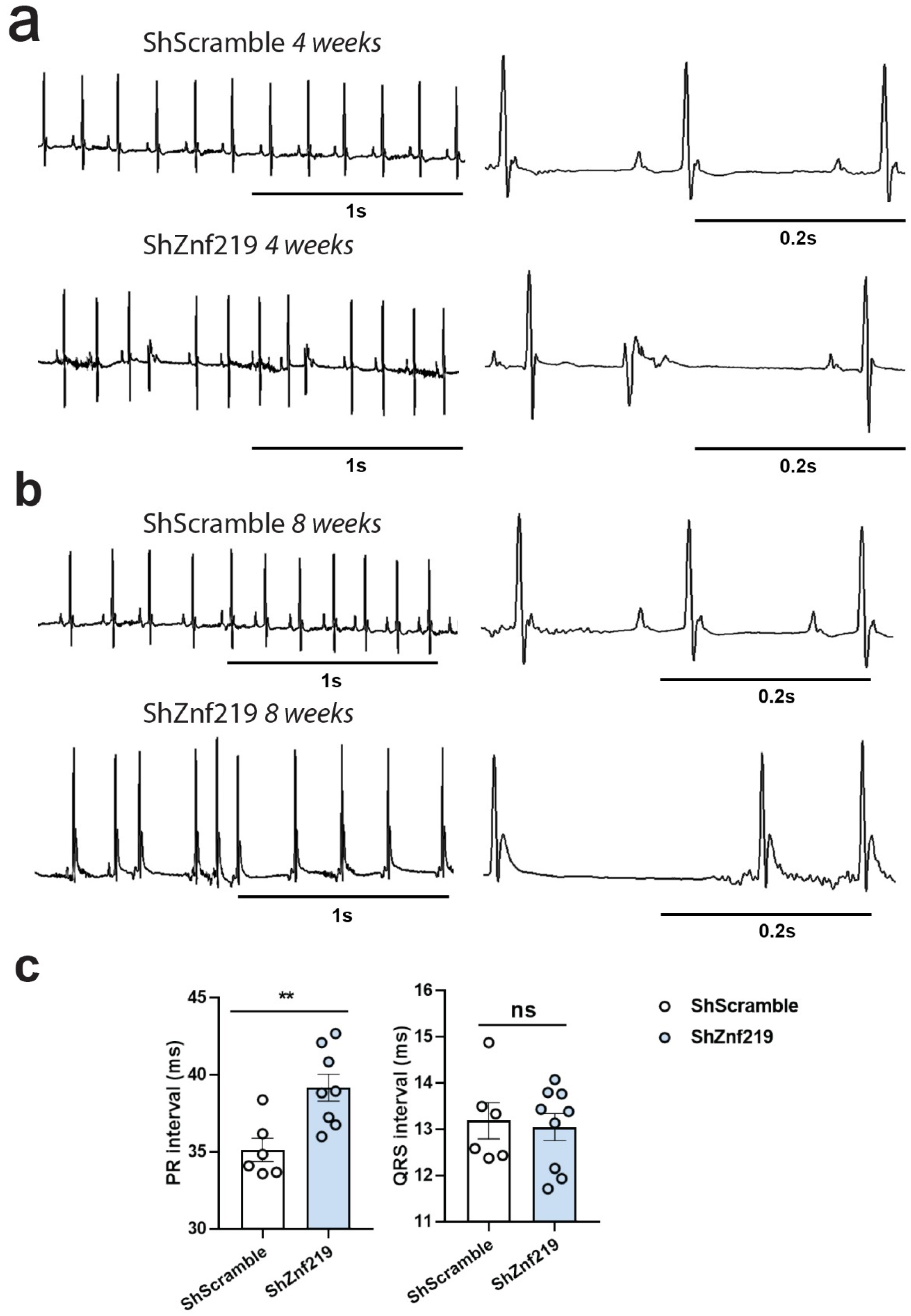
2.5. Cardiac Znf219 Knockdown Induces Arrhythmia in Adult Mice
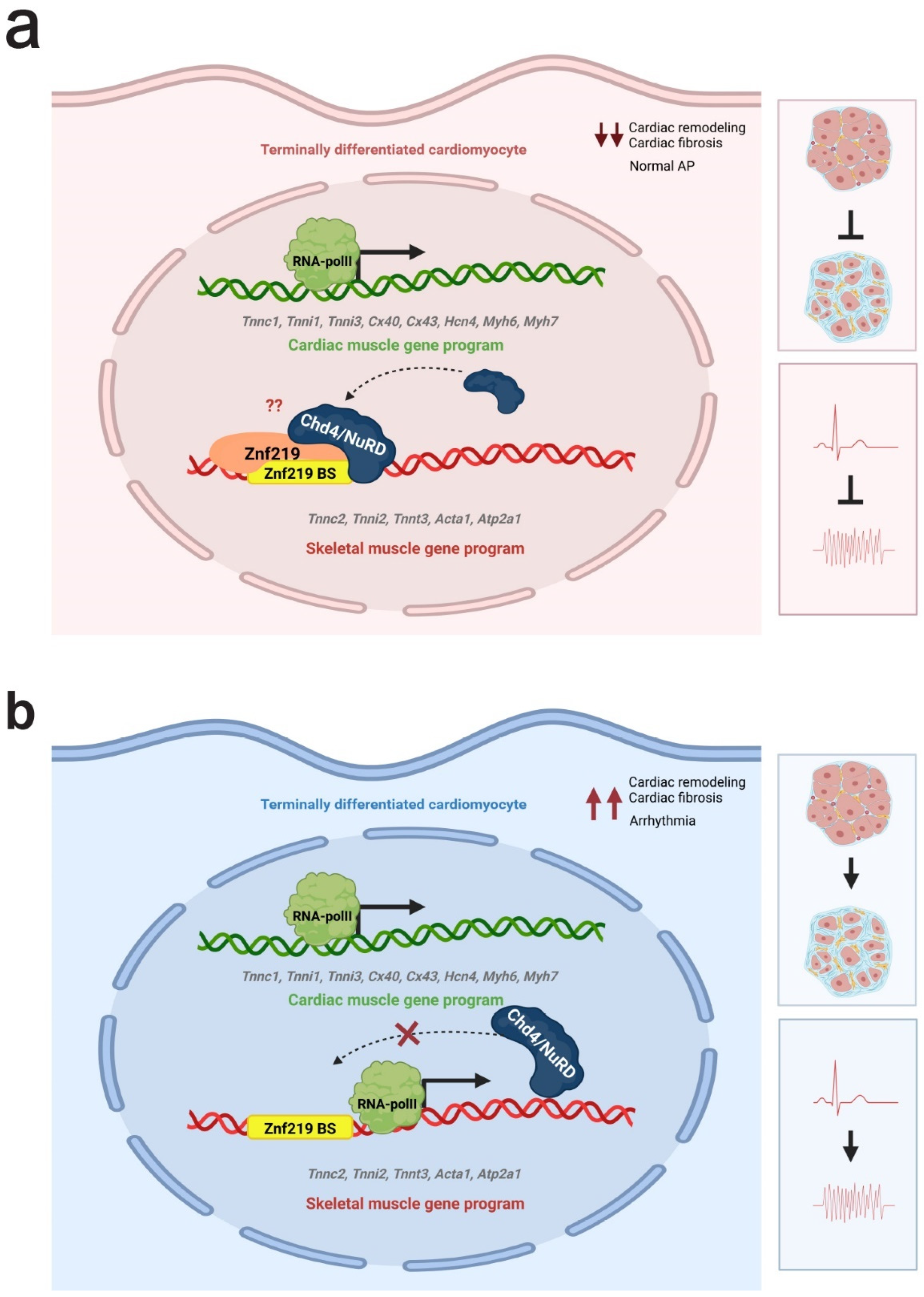
3. Discussion
Study Strengths and Limitations
4. Materials and Methods
4.1. Mouse Strains and Adeno-Associated Viral Infections
4.2. AAV9 Generation
4.3. Electrocardiography (ECG)
4.4. Protein Identification Using Mass Spectrometry
4.5. Cell Culture, Lentiviral Production, and Cell Infection
4.6. Transient Transfection with Calcium Phosphate
4.7. Reverse Transcription-qPCR (RT-PCR) and Gene Expression Analysis
4.8. Western Blotting
4.9. Histological Analysis
4.10. Transcription Factor (TF) Binding Analysis
4.11. Statistical Analysis and Software Used
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bauer, A.J.; Martin, K.A. Coordinating Regulation of Gene Expression in Cardiovascular Disease: Interactions between Chromatin Modifiers and Transcription Factors. Front. Cardiovasc. Med. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Finocchiaro, G.; Maurizi, N.; Crotti, L. Common presentation of rare cardiac diseases: Arrhythmias. Int. J. Cardiol. 2018, 257, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Kardiol. Pol. 2016, 74, 1359–1469. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.B.; Yin, X.; Gona, P.; Larson, M.G.; Beiser, A.S.; McManus, D.D.; Newton-Cheh, C.; Lubitz, S.A.; Magnani, J.W.; Ellinor, P.T.; et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: A cohort study. Lancet 2015, 386, 154–162. [Google Scholar] [CrossRef]
- Munshi, N.V. Gene regulatory networks in cardiac conduction system development. Circ. Res. 2012, 110, 1525–1537. [Google Scholar] [CrossRef]
- Park, D.S.; Fishman, G.I. Development and Function of the Cardiac Conduction System in Health and Disease. J. Cardiovasc. Dev. Dis. 2017, 4, 7. [Google Scholar] [CrossRef]
- Estrella, N.L.; Naya, F.J. Transcriptional networks regulating the costamere, sarcomere, and other cytoskeletal structures in striated muscle. Cell Mol. Life Sci. 2014, 71, 1641–1656. [Google Scholar] [CrossRef]
- Gomez-Del Arco, P.; Perdiguero, E.; Yunes-Leites, P.S.; Acin-Perez, R.; Zeini, M.; Garcia-Gomez, A.; Sreenivasan, K.; Jimenez-Alcazar, M.; Segales, J.; Lopez-Maderuelo, D.; et al. The Chromatin Remodeling Complex Chd4/NuRD Controls Striated Muscle Identity and Metabolic Homeostasis. Cell Metab. 2016, 23, 881–892. [Google Scholar] [CrossRef]
- Denslow, S.A.; Wade, P.A. The human Mi-2/NuRD complex and gene regulation. Oncogene 2007, 26, 5433–5438. [Google Scholar] [CrossRef]
- Wilczewski, C.M.; Hepperla, A.J.; Shimbo, T.; Wasson, L.; Robbe, Z.L.; Davis, I.J.; Wade, P.A.; Conlon, F.L. CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc. Natl. Acad. Sci. USA 2018, 115, 6727–6732. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Otero, M.G.; Grand, K.; Choi, A.; Graham, J.M., Jr.; Young, J.I.; Mackay, J.P. The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Sifrim, A.; Hitz, M.P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sif, S.; Jones, B.; Jackson, A.; Koipally, J.; Heller, E.; Winandy, S.; Viel, A.; Sawyer, A.; Ikeda, T.; et al. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 1999, 10, 345–355. [Google Scholar] [CrossRef]
- Naito, T.; Gomez-Del Arco, P.; Williams, C.J.; Georgopoulos, K. Antagonistic interactions between Ikaros and the chromatin remodeler Mi-2beta determine silencer activity and Cd4 gene expression. Immunity 2007, 27, 723–734. [Google Scholar] [CrossRef]
- Williams, C.J.; Naito, T.; Arco, P.G.; Seavitt, J.R.; Cashman, S.M.; De Souza, B.; Qi, X.; Keables, P.; Von Andrian, U.H.; Georgopoulos, K. The chromatin remodeler Mi-2beta is required for CD4 expression and T cell development. Immunity 2004, 20, 719–733. [Google Scholar] [CrossRef]
- Brayer, K.J.; Kulshreshtha, S.; Segal, D.J. The protein-binding potential of C2H2 zinc finger domains. Cell Biochem. Biophys. 2008, 51, 9–19. [Google Scholar] [CrossRef]
- Brayer, K.J.; Segal, D.J. Keep your fingers off my DNA: Protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys. 2008, 50, 111–131. [Google Scholar] [CrossRef]
- Sakai, T.; Hino, K.; Wada, S.; Maeda, H. Identification of the DNA binding specificity of the human ZNF219 protein and its function as a transcriptional repressor. DNA Res. 2003, 10, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, Y.; Hata, K.; Muramatsu, S.; Amano, K.; Ono, K.; Wakabayashi, M.; Matsuda, A.; Takada, K.; Nishimura, R.; Yoneda, T. The transcription factor Znf219 regulates chondrocyte differentiation by assembling a transcription factory with Sox9. J. Cell Sci. 2010, 123, 3780–3788. [Google Scholar] [CrossRef] [PubMed]
- Lien, H.W.; Yang, C.H.; Cheng, C.H.; Liao, Y.F.; Han, Y.S.; Huang, C.J. Zinc finger protein 219-like (ZNF219L) and Sox9a regulate synuclein-gamma2 (sncgb) expression in the developing notochord of zebrafish. Biochem. Biophys. Res. Commun. 2013, 442, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Lien, H.W.; Yang, C.H.; Cheng, C.H.; Hung, C.C.; Liao, W.H.; Hwang, P.P.; Han, Y.S.; Huang, C.J. A novel zinc finger protein 219-like (ZNF219L) is involved in the regulation of collagen type 2 alpha 1a (col2a1a) gene expression in zebrafish notochord. Int. J. Biol. Sci. 2013, 9, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Sharifi Tabar, M.; Mackay, J.P.; Low, J.K.K. The stoichiometry and interactome of the Nucleosome Remodeling and Deacetylase (NuRD) complex are conserved across multiple cell lines. FEBS J. 2019, 286, 2043–2061. [Google Scholar] [CrossRef]
- Ding, J.; Lin, Z.Q.; Jiang, J.M.; Seidman, C.E.; Seidman, J.G.; Pu, W.T.; Wang, D.Z. Preparation of rAAV9 to Overexpress or Knockdown Genes in Mouse Hearts. J. Vis. Exp. 2016, e54787. [Google Scholar] [CrossRef]
- Robbe, Z.L.; Shi, W.; Wasson, L.K.; Scialdone, A.P.; Wilczewski, C.M.; Sheng, X.; Hepperla, A.J.; Akerberg, B.N.; Pu, W.T.; Cristea, I.M.; et al. CHD4 is recruited by GATA4 and NKX2-5 to repress noncardiac gene programs in the developing heart. Genes. Dev. 2022, 36, 468–482. [Google Scholar] [CrossRef]
- Kim, M.K.; McGarry, T.J.; Ó Broin, P.; Flatow, J.M.; Golden, A.A.; Licht, J.D. An integrated genome screen identifies the Wnt signaling pathway as a major target of WT1. Proc. Natl. Acad. Sci. USA 2009, 106, 11154–11159. [Google Scholar] [CrossRef]
- Molenaar, J.P.; Verhoeven, J.I.; Rodenburg, R.J.; Kamsteeg, E.J.; Erasmus, C.E.; Vicart, S.; Behin, A.; Bassez, G.; Magot, A.; Pereon, Y.; et al. Clinical, morphological and genetic characterization of Brody disease: An international study of 40 patients. Brain 2020, 143, 452–466. [Google Scholar] [CrossRef]
- Ren, A.J.; Chen, C.; Zhang, S.; Liu, M.; Wei, C.; Wang, K.; Ma, X.; Song, Y.; Wang, R.; Zhang, H.; et al. Zbtb20 deficiency causes cardiac contractile dysfunction in mice. FASEB J. 2020, 34, 13862–13876. [Google Scholar] [CrossRef]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; Garcia-Prieto, J.; Garcia-Ruiz, J.M.; Pizarro, G.; Jimenez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruiz-Cabello, J.; et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, J.; Samulski, R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Guerra, A.; Roche-Molina, M.; Garcia-Quintans, N.; Sanchez-Ramos, C.; Martin-Perez, D.; Lytvyn, M.; de Nicolas-Hernandez, J.; Rivera-Torres, J.; Arroyo, D.F.; Sanz-Rosa, D.; et al. Sustained Elevated Blood Pressure Accelerates Atherosclerosis Development in a Preclinical Model of Disease. Int. J. Mol. Sci. 2021, 22, 8448. [Google Scholar] [CrossRef]
- Echevarría-Zomeño, S.; Fernández-Calviño, L.; Castro-Sanz, A.B.; López, J.A.; Vazquez, J.; Castellano, M.M. Dissecting the proteome dynamics of the early heat stress response leading to plant survival or death in Arabidopsis. Plant Cell Environ. 2016, 39, 1264–1278. [Google Scholar] [CrossRef]
- Martinez-Bartolome, S.; Navarro, P.; Martin-Maroto, F.; Lopez-Ferrer, D.; Ramos-Fernandez, A.; Villar, M.; Garcia-Ruiz, J.P.; Vazquez, J. Properties of average score distributions of SEQUEST: The probability ratio method. Mol. Cell Proteom. 2008, 7, 1135–1145. [Google Scholar] [CrossRef]
- Bonzon-Kulichenko, E.; Garcia-Marques, F.; Trevisan-Herraz, M.; Vazquez, J. Revisiting peptide identification by high-accuracy mass spectrometry: Problems associated with the use of narrow mass precursor windows. J. Proteome Res. 2015, 14, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Schones, D.E.; Smith, A.D.; Zhang, M.Q. Statistical significance of cis-regulatory modules. BMC Bioinform. 2007, 8, 19. [Google Scholar] [CrossRef]
- Matys, V.; Fricke, E.; Geffers, R.; Gossling, E.; Haubrock, M.; Hehl, R.; Hornischer, K.; Karas, D.; Kel, A.E.; Kel-Margoulis, O.V.; et al. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31, 374–378. [Google Scholar] [CrossRef]
- Perez-Llamas, C.; Lopez-Bigas, N. Gitools: Analysis and visualisation of genomic data using interactive heat-maps. PLoS ONE 2011, 6, e19541. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. Quantitative trait Loci analysis using the false discovery rate. Genetics 2005, 171, 783–790. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Oligo Forward (Fw) | Oligo Reverse (Rv) |
|---|---|---|
| m36B4 (Rplp0) | GCGACCTGGAAGTCCAACTA | ATCTGCTGCATCTGCTTGG |
| Znf219 | GGGACCCAGGCGAGATCC | CACGGCCCGCTTCAGATC |
| Chd4 | CTGCTCAGCGGGAGTGAGG | GGGTCTCGAGGTTTCTTAGGCT |
| Tnni2 | CACCTGAAGAGTGTGATGCTC | GGGCAGTGTTCTGACAGGTAG |
| Acta1 | GTGACCACAGCTGAACGTG | CCAGGGAGGAGGAAGAGG |
| Tnnc2 | GAGATGATCGCTGAGTTCAAG | GTCTGCCCTAGCATCCTCAT |
| Atp2a1 | TTTGGCAGGAACGGAATG | AGCCTTGATCCTTTGCACTG |
| Tnnt3 | ACAGATTGGCGGAGGAGAAG | CATGGAGGACAGAGCCTTTT |
| Cx40 | GGAGGAAAGGAAGCAGAAGG | GACTGTGGAGTGCTTGTGGA |
| Cacna1h | ATTTCTGGCCGAACAGTCC | AGCATCTTGGAGGCCTTTG |
| Hcn4 | TGACTTCAGATTTTACTGGGA | TTGAAGACGATCCAGGGTGT |
| Kcnh2 | TCGCTTTCTCAGGTTTCCCA | GCCTGGATCTGAGCCATGT |
| Scn5a | TCGTCATGGCATACACAACT | GTCTTCAGGCCTGAAATGA |
| Scn10a | TTGACAACTTCAATCAACAGAA | TGGTACTTATTCAAAGGCCGT |
| Col1a1 | GCTCCTCTTAGGGGCCACT | CCACGTCTCACCATTGGGG |
| Col3a1 | CTGTAACATGGAAACTGGGGAAA | CCATAGCTGAACTGAAAACCACC |
| Antibody | Host | Dilution | Secondary | Dilution |
|---|---|---|---|---|
| α-Flag | Mouse | 1:500 | α-mouse | 1:5000 or 1:3000 |
| α-Myc | Mouse | 1:15 | α-mouse | 1:5000 |
| α-tubulina | Mouse | 1:400,000 | α-mouse | 1:5000 |
| α-CHD4 | Rabbit | 1:500 | α-rabbit | 1:5000 |
| α-Znf219 | Mouse | 1:500 | α-mouse | 1:2000 |
| α-Tnni2 | Rabbit | 1:500 | α-rabbit | 1:3000 |
| α-Tnnt3 | Mouse | 1:500 | α-mouse | 1:3000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Abdellaoui-Soussi, F.; Yunes-Leites, P.S.; López-Maderuelo, D.; García-Marqués, F.; Vázquez, J.; Redondo, J.M.; Gómez-del Arco, P. Interplay between the Chd4/NuRD Complex and the Transcription Factor Znf219 Controls Cardiac Cell Identity. Int. J. Mol. Sci. 2022, 23, 9565. https://doi.org/10.3390/ijms23179565
El Abdellaoui-Soussi F, Yunes-Leites PS, López-Maderuelo D, García-Marqués F, Vázquez J, Redondo JM, Gómez-del Arco P. Interplay between the Chd4/NuRD Complex and the Transcription Factor Znf219 Controls Cardiac Cell Identity. International Journal of Molecular Sciences. 2022; 23(17):9565. https://doi.org/10.3390/ijms23179565
Chicago/Turabian StyleEl Abdellaoui-Soussi, Fadoua, Paula S. Yunes-Leites, Dolores López-Maderuelo, Fernando García-Marqués, Jesús Vázquez, Juan Miguel Redondo, and Pablo Gómez-del Arco. 2022. "Interplay between the Chd4/NuRD Complex and the Transcription Factor Znf219 Controls Cardiac Cell Identity" International Journal of Molecular Sciences 23, no. 17: 9565. https://doi.org/10.3390/ijms23179565
APA StyleEl Abdellaoui-Soussi, F., Yunes-Leites, P. S., López-Maderuelo, D., García-Marqués, F., Vázquez, J., Redondo, J. M., & Gómez-del Arco, P. (2022). Interplay between the Chd4/NuRD Complex and the Transcription Factor Znf219 Controls Cardiac Cell Identity. International Journal of Molecular Sciences, 23(17), 9565. https://doi.org/10.3390/ijms23179565