
Enhancing the Catalytic Activity of Type II L-Asparaginase from Bacillus licheniformis through Semi-Rational Design


 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
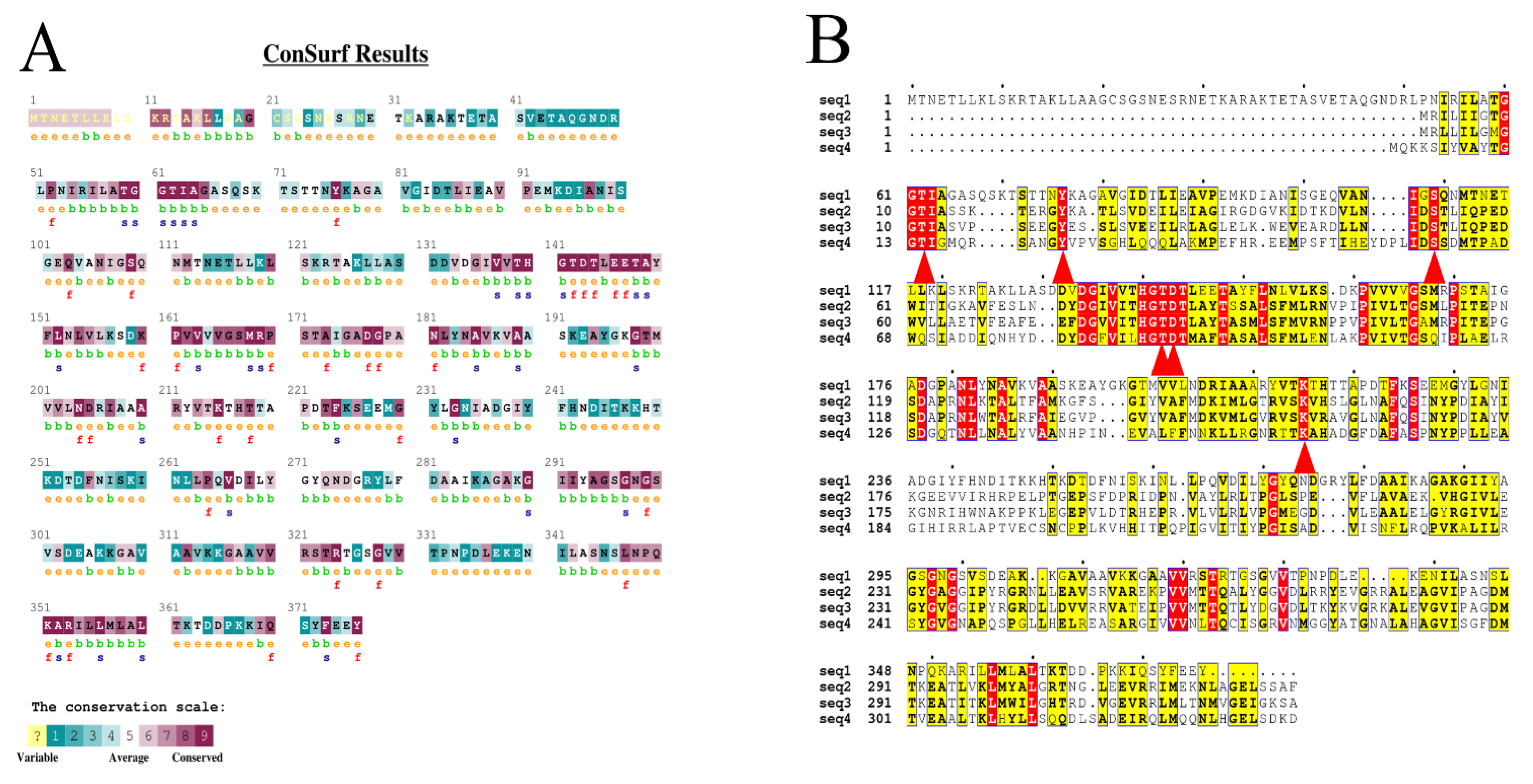
2.1. Generation of Mutation Sites
2.2. Screening of Mutant Libraries
2.3. Enzymatic Characterization of Wild-Type and Mutant ILRAC
2.4. Structure Stability Analysis of Wild-Type and Mutant ILRAC
2.5. Substrate Affinity Analysis of Wild-Type and Mutant ILRAC
3. Materials and Methods
3.1. Strains and Reagents
3.2. Bioinformatics Analysis and Selection of Mutation Sites
3.3. Saturation Mutagenesis
3.4. Construction of Combination Mutants
3.5. Protein Expression and Purification of BliansA
3.6. L-Asparaginase Activity Assay
3.7. Determination of Kinetic Parameters
3.8. Effect of pH and Temperature on Enzyme Activity and Stability
3.9. Circular Dichroism Spectroscopy Analysis
3.10. Molecular Dynamic (MD) Simulations
= ΔEinternal + ΔEVDW + ΔEelec + ΔGGB + ΔGSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nunes, J.C.F.; Cristovao, R.O.; Freire, M.G.; Santos-Ebinuma, V.C.; Faria, J.L.; Silva, C.G.; Tavares, A.P. Recent Strategies and Applications for l-Asparaginase Confinement. Molecules 2020, 25, 5827. [Google Scholar] [CrossRef]
- Michalska, K.; Jaskolski, M. Structural aspects of L-asparaginases, their friends and relations. Acta Biochim. Pol. 2006, 53, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Chand, S.; Mahajan, R.V.; Prasad, J.P.; Sahoo, D.K.; Mihooliya, K.N.; Dhar, M.S.; Sharma, G. A comprehensive review on microbial l-asparaginase: Bioprocessing, characterization, and industrial applications. Biotechnol. Appl. Biochem. 2020, 67, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Batool, T.; Makky, E.A.; Jalal, M.; Yusoff, M.M. A Comprehensive Review on l-Asparaginase and Its Applications. Appl. Biochem. Biotechnol. 2015, 178, 900–923. [Google Scholar] [CrossRef] [PubMed]
- Aisha, A.; Zia, M.A.; Asger, M.; Muhammad, F. L-asparaginase, acrylamide quenching enzyme production from leaves of Tamarindus indica and seeds of Vigna radiata–Fabaceae. Pak. J. Bot. 2020, 52, 52. [Google Scholar] [CrossRef]
- Xu, F.; Oruna-Concha, M.-J.; Elmore, J.S. The use of asparaginase to reduce acrylamide levels in cooked food. Food Chem. 2016, 210, 163–171. [Google Scholar] [CrossRef]
- Jiao, L.; Chi, H.; Lu, Z.; Zhang, C.; Chia, S.R.; Show, P.L.; Tao, Y.; Lu, F. Characterization of a novel type I l-asparaginase from Acinetobacter soli and its ability to inhibit acrylamide formation in potato chips. J. Biosci. Bioeng. 2020, 129, 672–678. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Wu, H.; Zhang, W.; Guang, C.; Mu, W. Microbial production, molecular modification, and practical application of l-Asparaginase: A review. Int. J. Biol. Macromol. 2021, 186, 975–983. [Google Scholar] [CrossRef]
- Gu, S.; Dai, X.; Jiang, J.; Liu, Y. Enhancing the catalytic activity of cyanobacterial chlorophyllase from Oscillatoria acuminata PCC 6304 through rational site-directed mutagenesis. Process Biochem. 2022, 114, 111–118. [Google Scholar] [CrossRef]
- Sequeiros-Borja, C.E.; Surpeta, B.; Brezovsky, J. Recent advances in user-friendly computational tools to engineer protein function. Briefings Bioinform. 2020, 22, bbaa150. [Google Scholar] [CrossRef]
- Karamitros, C.S.; Konrad, M. Fluorescence-Activated Cell Sorting of Human l-asparaginase Mutant Libraries for Detecting Enzyme Variants with Enhanced Activity. ACS Chem. Biol. 2016, 11, 2596–2607. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Zhang, X.; Rao, Z.; Chen, K.; Xu, M.; Yang, T.; Yang, S. Amino acid residues adjacent to the catalytic cavity of tetramer l-asparaginase II contribute significantly to its catalytic efficiency and thermostability. Enzym. Microb. Technol. 2016, 82, 15–22. [Google Scholar] [CrossRef]
- Barnsley, K.K.; Ondrechen, M.J. Enzyme active sites: Identification and prediction of function using computational chemistry. Curr. Opin. Struct. Biol. 2022, 74, 102384. [Google Scholar] [CrossRef] [PubMed]
- Zajac, J.; Anderson, H.; Adams, L.; Wangmo, D.; Suhail, S.; Almen, A.; Berns, L.; Coerber, B.; Dawson, L.; Hunger, A.; et al. Effects of Distal Mutations on Prolyl-Adenylate Formation of Escherichia coli Prolyl-tRNA Synthetase. Protein J. 2020, 39, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Ngu, L.; Winters, J.N.; Nguyen, K.; Ramos, K.E.; DeLateur, N.A.; Makowski, L.; Whitford, P.C.; Ondrechen, M.J.; Beuning, P.J. Probing remote residues important for catalysis in Escherichia coli ornithine transcarbamoylase. PLoS ONE 2020, 15, e0228487. [Google Scholar] [CrossRef]
- Gu, J.; Sim, B.R.; Li, J.; Yu, Y.; Qin, L.; Wu, L.; Shen, Y.; Nie, Y.; Zhao, Y.-L.; Xu, Y. Evolutionary coupling-inspired engineering of alcohol dehydrogenase reveals the influence of distant sites on its catalytic efficiency for stereospecific synthesis of chiral alcohols. Comput. Struct. Biotechnol. J. 2021, 19, 5864–5873. [Google Scholar] [CrossRef]
- Sumbalova, L.; Stourac, J.; Martinek, T.; Bednar, D.; Damborsky, J. HotSpot Wizard 3.0: Web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res. 2018, 46, W356–W362. [Google Scholar] [CrossRef]
- Kwiatos, N.; Jędrzejczak-Krzepkowska, M.; Krzemińska, A.; Delavari, A.; Paneth, P.; Bielecki, S. Evolved Fusarium oxysporum laccase expressed in Saccharomyces cerevisiae. Sci. Rep. 2020, 10, 3244. [Google Scholar] [CrossRef]
- Zhang, L.; Singh, R.; Sivakumar, D.; Guo, Z.; Li, J.; Chen, F.; He, Y.; Guan, X.; Kang, Y.C.; Lee, J.-K. An artificial synthetic pathway for acetoin, 2,3-butanediol, and 2-butanol production from ethanol using cell free multi-enzyme catalysis. Green Chem. 2017, 20, 230–242. [Google Scholar] [CrossRef]
- Jamek, S.B.; Muschiol, J.; Holck, J.; Zeuner, B.; Busk, P.K.; Mikkelsen, J.D.; Meyer, A.S. Loop Protein Engineering for Improved Transglycosylation Activity of a β- N -Acetylhexosaminidase. ChemBioChem 2018, 19, 1858–1865. [Google Scholar] [CrossRef]
- Marks, D.S.; Colwell, L.J.; Sheridan, R.; Hopf, T.A.; Pagnani, A.; Zecchina, R.; Sander, C. Protein 3D Structure Computed from Evolutionary Sequence Variation. PLoS ONE 2011, 6, e28766. [Google Scholar] [CrossRef]
- Pan, B.; Akyuz, N.; Liu, X.-P.; Asai, Y.; Nist-Lund, C.; Kurima, K.; Derfler, B.H.; György, B.; Limapichat, W.; Walujkar, S.; et al. TMC1 Forms the Pore of Mechanosensory Transduction Channels in Vertebrate Inner Ear Hair Cells. Neuron 2018, 99, 736–753.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jing, X.; Deng, Y.; Nie, Y.; Xu, F.; Xu, Y.; Zhao, Y.; Hunt, J.F.; Montelione, G.T.; Szyperski, T. Evolutionary coupling saturation mutagenesis: Coevolution-guided identification of distant sites influencing Bacillus naganoensis pullulanase activity. FEBS Lett. 2019, 594, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Lv, K.; Li, J.; Wu, B.; He, B. Coevolutionary analysis reveals a distal amino acid residue pair affecting the catalytic activity of GH5 processive endoglucanase from Bacillus subtilis BS-5. Biotechnol. Bioeng. 2022, 119, 2105–2114. [Google Scholar] [CrossRef]
- Ma, D.; Xin, Y.; Guo, Z.; Shi, Y.; Zhang, L.; Li, Y.; Gu, Z.; Ding, Z.; Shi, G. Ancestral sequence reconstruction and spatial structure analysis guided alteration of longer-chain substrate catalysis for Thermomicrobium roseum lipase. Enzym. Microb. Technol. 2022, 156, 109989. [Google Scholar] [CrossRef]
- Ding, Z.; Ahmed, S.; Hang, J.; Mi, H.; Hou, X.; Yang, G.; Huang, Z.; Lu, X.; Zhang, W.; Liu, S.; et al. Rationally engineered chitin deacetylase from Arthrobacter sp. AW19M34-1 with improved catalytic activity toward crystalline chitin. Carbohydr. Polym. 2021, 274, 118637. [Google Scholar] [CrossRef]
- Sudhir, A.P.; Agarwaal, V.V.; Dave, B.R.; Patel, D.H.; Subramanian, R. Enhanced catalysis of l-asparaginase from Bacillus licheniformis by a rational redesign. Enzym. Microb. Technol. 2016, 86, 1–6. [Google Scholar] [CrossRef]
- Chi, H.; Chen, M.; Jiao, L.; Lu, Z.; Bie, X.; Zhao, H.; Lu, F. Characterization of a Novel L-Asparaginase from Mycobacterium gordonae with Acrylamide Mitigation Potential. Foods 2021, 10, 2819. [Google Scholar] [CrossRef]
- Vidya, J.; Ushasree, M.V.; Pandey, A. Effect of surface charge alteration on stability of l-asparaginase II from Escherichia sp. Enzym. Microb. Technol. 2014, 56, 15–19. [Google Scholar] [CrossRef]
- Kotzia, G.A.; Labrou, N.E. Engineering thermal stability of l-asparaginase by in vitro directed evolution. FEBS J. 2009, 276, 1750–1761. [Google Scholar] [CrossRef]
- Beckett, A.; Gervais, D. What makes a good new therapeutic L-asparaginase? World J. Microbiol. Biotechnol. 2019, 35, 152. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, J.; Jiao, L.; Zhong, L.; Lu, Z.; Zhang, C.; Lu, F. Improvement of the activity of l-asparaginase I improvement of the catalytic activity of l-asparaginase I from Bacillus megaterium H-1 by in vitro directed evolution. J. Biosci. Bioeng. 2019, 128, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liu, S.; Pang, C.; Gao, H.; Wang, M.; Du, G. Improvement of catalytic efficiency and thermal stability of l-asparaginase from Bacillus subtilis 168 through reducing the flexibility of the highly flexible loop at N-terminus. Process Biochem. 2019, 78, 42–49. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, S.; Jiao, Y.; Gao, H.; Wang, M.; Du, G.; Chen, J. Enhanced extracellular production of L-asparaginase from Bacillus subtilis 168 by B. subtilis WB600 through a combined strategy. Appl. Microbiol. Biotechnol. 2016, 101, 1509–1520. [Google Scholar] [CrossRef]
- Alrumman, S.A.; Mostafa, Y.S.; Al-Izran, K.A.; Alfaifi, M.Y.; Taha, T.H.; Elbehairi, S.E. Production and Anticancer Activity of an L-Asparaginase from Bacillus licheniformis Isolated from the Red Sea, Saudi Arabia. Sci. Rep. 2019, 9, 3756. [Google Scholar] [CrossRef]
- Qeshmi, F.I.; Homaei, A.; Khajeh, K.; Kamrani, E.; Fernandes, P. Production of a Novel Marine Pseudomonas aeruginosa Recombinant L-Asparaginase: Insight on the Structure and Biochemical Characterization. Mar. Biotechnol. 2022, 24, 599–613. [Google Scholar] [CrossRef]
- El-Naggar, N.E.-A.; Deraz, S.F.; Soliman, H.M.; El-Deeb, N.M.; El-Ewasy, S.M. Purification, characterization, cytotoxicity and anticancer activities of L-asparaginase, anti-colon cancer protein, from the newly isolated alkaliphilic Streptomyces fradiae NEAE-82. Sci. Rep. 2016, 6, 32926. [Google Scholar] [CrossRef]
- Yuen, C.M.; Liu, D.R. Dissecting protein structure and function using directed evolution. Nat. Chem. Biol. 2007, 4, 995–997. [Google Scholar] [CrossRef]
- Wong, K.-S.; Fong, W.-P.; Tsang, P.W.-K. A single Phe54Tyr substitution improves the catalytic activity and thermostability of Trigonopsis variabilis D-amino acid oxidase. New Biotechnol. 2010, 27, 78–84. [Google Scholar] [CrossRef]
- Ardalan, N.; Sepahi, A.A.; Khavari-Nejad, R.A. Development of Escherichia coli asparaginase II for the Treatment of Acute Lymphocytic Leukemia: In Silico Reduction of asparaginase II Side Effects by a Novel Mutant (V27F). Asian Pac. J. Cancer Prev. 2021, 22, 1137–1147. [Google Scholar] [CrossRef]
- Aghaeepoor, M.; Akbarzadeh, A.; Mirzaie, S.; Hadian, A.; Aval, S.J.; Dehnavi, E. Selective reduction in glutaminase activity of l-Asparaginase by asparagine 248 to serine mutation: A combined computational and experimental effort in blood cancer treatment. Int. J. Biol. Macromol. 2018, 120, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Eckes, K.M.; Mu, X.; Ruehle, M.A.; Ren, P.; Suggs, L.J. β sheets not required: Combined experimental and computational studies of self-assembly and gelation of the ester-containing analogue of an Fmoc-dipeptide hydrogelator. Langmuir 2014, 30, 5287–5296. [Google Scholar] [CrossRef] [PubMed]
- Farshadfar, C.; Mollica, A.; Rafii, F.; Noorbakhsh, A.; Nikzad, M.; Seyedi, S.H.; Abdi, F.; Verki, S.A.; Mirzaie, S. Staphylococcus aureus. Mol. Simul. 2020, 46, 507–520. [Google Scholar] [CrossRef]
- Rahman, M.M.; Saha, T.; Islam, K.J.; Suman, R.H.; Biswas, S.; Rahat, E.U.; Hossen, M.R.; Islam, R.; Hossain, M.N.; Mamun, A.A.; et al. Virtual screening, molecular dynamics and structure-activity relationship studies to identify potent approved drugs for COVID-19 treatment. J. Biomol. Struct. Dyn. 2021, 39, 6231–6241. [Google Scholar] [CrossRef]
- Aghaiypour, K.; Wlodawer, A.A.; Lubkowski, J. Structural Basis for the Activity and Substrate Specificity of Erwinia chrysanthemi l-Asparaginase. Biochemistry 2001, 40, 5655–5664. [Google Scholar] [CrossRef]
- Ran, T.; Jiao, L.; Wang, W.; Chen, J.; Chi, H.; Lu, Z.; Zhang, C.; Xu, D.; Lu, F. Structures of l-asparaginase from Bacillus licheniformis Reveal an Essential Residue for its Substrate Stereoselectivity. J. Agric. Food Chem. 2020, 69, 223–231. [Google Scholar] [CrossRef]
- Offman, M.N.; Krol, M.; Patel, N.; Krishnan, S.; Liu, J.; Saha, V.; Bates, P.A. Rational engineering of L-asparaginase reveals importance of dual activity for cancer cell toxicity. Blood 2011, 117, 1614–1621. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Y.; Wang, J.; Yu, Z.; Liu, Y.; Tong, Y.; Han, W. Adaptive Steered Molecular Dynamics Combined with Protein Structure Networks Revealing the Mechanism of Y68I/G109P Mutations That Enhance the Catalytic Activity of D-psicose 3-Epimerase from Clostridium Bolteae. Front. Chem. 2018, 6, 437. [Google Scholar] [CrossRef]
- Yin, C.; Zheng, T.; Chang, X. Biosynthesis of S-Adenosylmethionine by Magnetically Immobilized Escherichia coli Cells Highly Expressing a Methionine Adenosyltransferase Variant. Molecules 2017, 22, 1365. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, Y.; Wu, M.; Zhu, L.; Yang, L.; Lin, J. Semi-rationally engineered variants of S-adenosylmethionine synthetase from Escherichia coli with reduced product inhibition and improved catalytic activity. Enzym. Microb. Technol. 2019, 129, 109355. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Srivastava, A.; Mukherjee, G.; Pandey, R.; Verma, A.K.; Mishra, P.; Kundu, B. Hyperthermophilic asparaginase mutants with enhanced substrate affinity and antineoplastic activity: Structural insights on their mechanism of action. FASEB J. 2011, 26, 1161–1171. [Google Scholar] [CrossRef]
- Yim, S.; Kim, M. Purification and characterization of thermostable l-asparaginase from Bacillus amyloliquefaciens MKSE in Korean soybean paste. LWT 2019, 109, 415–421. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef]
- Wang, J.M.; Wang, W.; Kollman, P.A. Antechamber: An accessory software package for molecular mechanical calculations. Abstr. Pap. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Schöning-Stierand, K.; Diedrich, K.; Ehrt, C.; Flachsenberg, F.; Graef, J.; Sieg, J.; Penner, P.; Poppinga, M.; Ungethüm, A.; Rarey, M. ProteinsPlus: A comprehensive collection of web-based molecular modeling tools. Nucleic Acids Res. 2022, 50, W611–W615. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Vmax (mmol·min−1·mg−1) | Km (mM) | kcat (min−1) | kcat/Km (min−1·mM−1) |
|---|---|---|---|---|
| WT | 9.93 ± 0.94 | 2.33 ± 0.49 | 197.95 ± 5.42 | 84.89 |
| ILRAC | 11.42 ± 1.63 | 1.45 ± 0.18 | 778.87 ± 36.43 | 537.15 |
| Secondary Structure | Proportion (%) | |
|---|---|---|
| WT | ILRAC | |
| α-Helices | 26.0 | 34.5 |
| β-Sheets | 16.9 | 11.0 |
| Turns | 17.5 | 23.1 |
| Random coli | 39.6 | 31.4 |
| Enzymes | Hydrogen Bonds Number | Rg (Å) | SASA (Å2) |
|---|---|---|---|
| WT | 810.63 ± 28.92 | 20.35 ± 0.08 | 36,276.87 ± 1323.39 |
| ILRAC | 812.60 ± 26.58 | 20.38 ± 0.07 | 36,675.61 ± 968.37 |
| System Name | WT-ASN | ILRAC-ASN |
|---|---|---|
| ΔEvdw 1 | −12.24 ± 3.34 | −16.57 ± 2.85 |
| ΔEelec 2 | −128.76 ± 5.85 | −126.43 ± 7.69 |
| ΔGGB 3 | 111.02 ± 6.67 | 110.95 ± 6.18 |
| ΔGSA 4 | −2.87 ± 0.09 | −3.17 ± 0.09 |
| ΔGbind 5 | −32.86 ± 2.48 | −35.23 ± 2.52 |
| WT | ILRAC | |
|---|---|---|
| volume (Å3) | 1216.88 | 1248.40 |
| surface (Å2) | 1074.33 | 1100.43 |
| depth (Å) | 28.61 | 28.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Jiao, L.; Shen, J.; Chi, H.; Lu, Z.; Liu, H.; Lu, F.; Zhu, P. Enhancing the Catalytic Activity of Type II L-Asparaginase from Bacillus licheniformis through Semi-Rational Design. Int. J. Mol. Sci. 2022, 23, 9663. https://doi.org/10.3390/ijms23179663
Zhou Y, Jiao L, Shen J, Chi H, Lu Z, Liu H, Lu F, Zhu P. Enhancing the Catalytic Activity of Type II L-Asparaginase from Bacillus licheniformis through Semi-Rational Design. International Journal of Molecular Sciences. 2022; 23(17):9663. https://doi.org/10.3390/ijms23179663
Chicago/Turabian StyleZhou, Yawen, Linshu Jiao, Juan Shen, Huibing Chi, Zhaoxin Lu, Huawei Liu, Fengxia Lu, and Ping Zhu. 2022. "Enhancing the Catalytic Activity of Type II L-Asparaginase from Bacillus licheniformis through Semi-Rational Design" International Journal of Molecular Sciences 23, no. 17: 9663. https://doi.org/10.3390/ijms23179663
APA StyleZhou, Y., Jiao, L., Shen, J., Chi, H., Lu, Z., Liu, H., Lu, F., & Zhu, P. (2022). Enhancing the Catalytic Activity of Type II L-Asparaginase from Bacillus licheniformis through Semi-Rational Design. International Journal of Molecular Sciences, 23(17), 9663. https://doi.org/10.3390/ijms23179663