
Variable Expression of GABAA Receptor Subunit Gamma 2 Mutation in a Nuclear Family Displaying Developmental and Encephalopathic Phenotype

 , ,
, , 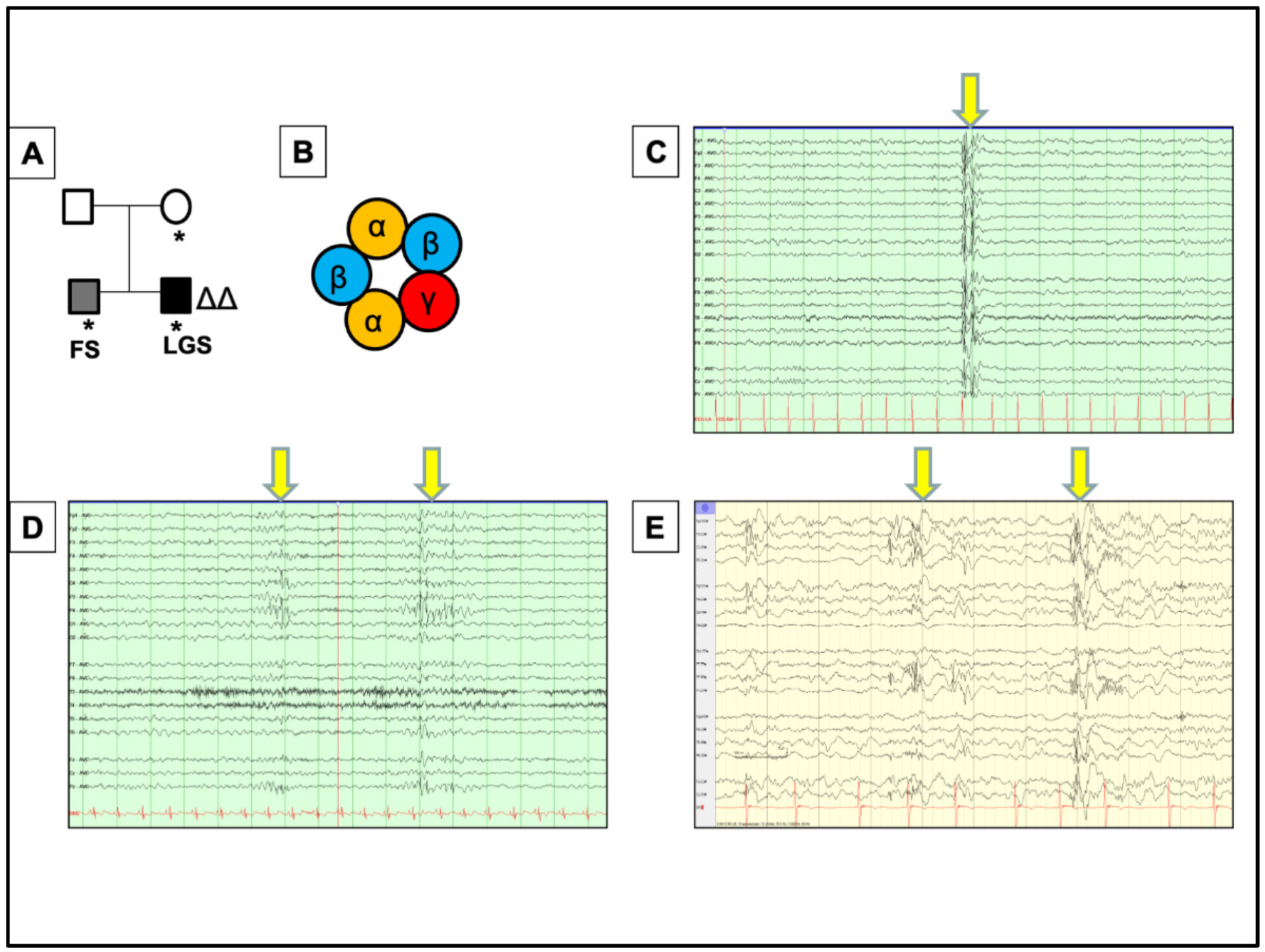{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. A Novel Case of GABRG2 Mutation Associated with DEE, Expanding the Disease Phenotypes Associated with GABRG2 in DEEs
3. Unique Functional Properties of GABRG2-Encoded γ2 Subunit
4. Pathomechanisms for GABRG2 Mutations in DEEs
5. Unique Functional Properties of GABRB3 Encoded β3 Subunit
6. Pathomechanisms for GABRB3 Mutations in DEEs
7. Non-GABR Mutations Associated with DEEs and Therapeutic Interventions
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheffer, I.E.; Liao, J. Deciphering the concepts behind “Epileptic encephalopathy” and “Developmental and epileptic encephalopathy”. Eur. J. Paediatr. Neurol. 2020, 24, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Curatolo, P. Developmental and epileptic encephalopathies: What we do and do not know. Brain 2020, 144, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Meena; Aznarez, I.; et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020, 12, eaaz6100. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Santiago, L.; Isom, L.L. Dravet Syndrome: A Developmental and Epileptic Encephalopathy. Epilepsy Curr. 2019, 19, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Gorman, K.M.; Peters, C.H.; Lynch, B.; Jones, L.; Bassett, D.S.; King, M.D.; Ruben, P.C.; Rosch, R.E. Persistent sodium currents in SCN1A developmental and degenerative epileptic dyskinetic encephalopathy. Brain Commun. 2021, 3, fcab235. [Google Scholar] [CrossRef]
- Camp, C.R.; Yuan, H. GRIN2D/GluN2D NMDA receptor: Unique features and its contribution to pediatric developmental and epileptic encephalopathy. Eur. J. Paediatr. Neurol. 2020, 24, 89–99. [Google Scholar] [CrossRef]
- Amador, A.; Bostick, C.D.; Olson, H.; Peters, J.; Camp, C.R.; Krizay, D.; Chen, W.; Han, W.; Tang, W.; Kanber, A.; et al. Modelling and treating GRIN2A developmental and epileptic encephalopathy in mice. Brain 2020, 143, 2039–2057. [Google Scholar] [CrossRef]
- Tsuchida, N.; Hamada, K.; Shiina, M.; Kato, M.; Kobayashi, Y.; Tohyama, J.; Kimura, K.; Hoshino, K.; Ganesan, V.; Teik, K.W.; et al. GRIN2D variants in three cases of developmental and epileptic encephalopathy. Clin. Genet. 2018, 94, 538–547. [Google Scholar] [CrossRef]
- Cherubini, E. Phasic GABAA-Mediated Inhibition. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Lee, V.; Maguire, J. The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front. Neural Circuits 2014, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M., Jr.; Kim, J.J.; Hibbs, R.E. Structure of a human synaptic GABAA receptor. Nature 2018, 559, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Lakhan, R.; Kalita, J.; Garg, R.; Misra, U.; Mittal, B. Potential role of GABAA receptor subunit; GABRA6, GABRB2 and GABRR2 gene polymorphisms in epilepsy susceptibility and pharmacotherapy in North Indian population. Clin. Chim. Acta Int. J. Clin. Chem. 2011, 412, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.C.; Macdonald, R.L. A structural look at GABAA receptor mutations linked to epilepsy syndromes. Brain Res. 2019, 1714, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Kang, J.Q.; Gallagher, M.J. GABAA Receptor Subunit Mutations and Genetic Epilepsies. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Wallace, R.H.; Marini, C.; Petrou, S.; Harkin, L.A.; Bowser, D.N.; Panchal, R.G.; Williams, D.A.; Sutherland, G.R.; Mulley, J.C.; Scheffer, I.E.; et al. Mutant GABAA receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001, 28, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.J.; Song, L.; Arain, F.; Macdonald, R.L. The Juvenile Myoclonic Epilepsy GABAA Receptor 1 Subunit Mutation A322D Produces Asymmetrical, Subunit Position-Dependent Reduction of Heterozygous Receptor Currents and 1 Subunit Protein Expression. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 5570–5578. [Google Scholar] [CrossRef] [PubMed]
- Harkin, L.A.; Bowser, D.N.; Dibbens, L.M.; Singh, R.; Phillips, F.; Wallace, R.H.; Richards, M.C.; Williams, D.A.; Mulley, J.C.; Berkovic, S.F.; et al. Truncation of the GABAA-Receptor γ2 Subunit in a Family with Generalized Epilepsy with Febrile Seizures Plus. Am. J. Hum. Genet. 2002, 70, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.C.; Tian, X.; Hu, N.; Shen, W.; Catron, M.A.; Yang, Y.; Chen, J.; Jiang, Y.; Zhang, Y.; Macdonald, R.L. Dravet syndrome-associated mutations in GABRA1, GABRB2 and GABRG2 define the genetic landscape of defects of GABAA receptors. Brain Commun. 2021, 3, fcab033. [Google Scholar] [CrossRef]
- Qu, S.; Catron, M.; Zhou, C.; Janve, V.; Shen, W.; Howe, R.K.; Macdonald, R.L. GABAA receptor β3 subunit mutation D120N causes Lennox–Gastaut syndrome in knock-in mice. Brain Commun. 2020, 2, fcaa028. [Google Scholar] [CrossRef]
- Tanaka, M.; Olsen, R.W.; Medina, M.T.; Schwartz, E.; Alonso, M.E.; Duron, R.M.; Castro-Ortega, R.; Martinez-Juarez, I.E.; Pascual-Castroviejo, I.; Machado-Salas, J.; et al. Hyperglycosylation and Reduced GABA Currents of Mutated GABRB3 Polypeptide in Remitting Childhood Absence Epilepsy. Am. J. Hum. Genet. 2008, 82, 1249–1261. [Google Scholar] [CrossRef]
- Huang, X.; Tian, M.; Hernandez, C.C.; Hu, N.; Macdonald, R.L. The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol. Dis. 2012, 48, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, R.L.; Kang, J.-Q.; Gallagher, M.J. Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 2010, 588 Pt 11, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Cossette, P.; Liu, L.; Brisebois, K.; Dong, H.; Lortie, A.; Vanasse, M.; Saint-Hilaire, J.-M.; Carmant, L.; Verner, A.; Lu, W.-Y.; et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 2002, 31, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Bouteiller, D.; Keren, B.; Cheuret, E.; Poirier, K.; Trouillard, O.; Benyahia, B.; Quelin, C.; Carpentier, W.; Julia, S.; et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009, 5, e1000381. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, X.D.; Zheng, G.; Zhang, L.J. Chemotherapy-induced brain changes in breast cancer survivors: Evaluation with multimodality magnetic resonance imaging. Brain Imaging Behav. 2019, 13, 1799–1814. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, C. The γ2 subunit of GABAA receptors is required for maintenance of receptors at mature synapses. Mol. Cell. Neurosci. 2003, 24, 442–450. [Google Scholar] [CrossRef]
- Essrich, C.; Lorez, M.; Benson, J.A.; Fritschy, J.-M.; Luscher, B. Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat. Neurosci. 1998, 1, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Alldred, M.J.; Mulder-Rosi, J.; Lingenfelter, S.E.; Chen, G.; Lüscher, B. Distinct 2 Subunit Domains Mediate Clustering and Synaptic Function of Postsynaptic GABAA Receptors and Gephyrin. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 594–603. [Google Scholar] [CrossRef]
- Angelotti, T.P.; Macdonald, R.L. Assembly of GABAA receptor subunits: Alpha 1 beta 1 and alpha 1 beta 1 gamma 2S subunits produce unique ion channels with dissimilar single- channel properties. J. Neurosci. Off. J. Soc. Neurosci. 1993, 13, 1429–1440. [Google Scholar] [CrossRef]
- Lorez, M.; Benke, D.; Lüscher, B.; Möhler, H.; Benson, J.A. Single-channel properties of neuronal GABA A receptors from mice lacking the γ2 subunit. J. Physiol. 2000, 527, 11–31. [Google Scholar] [CrossRef]
- Gurba, K.N.; Hernandez, C.; Hu, N.; Macdonald, R.L. GABRB3 Mutation, G32R, Associated with Childhood Absence Epilepsy Alters α1β3γ2L γ-Aminobutyric Acid Type A (GABAA) Receptor Expression and Channel Gating. J. Biol. Chem. 2012, 287, 12083–12097. [Google Scholar] [CrossRef] [Green Version]
- Günther, U.; Benson, J.; Benke, D.; Fritschy, J.M.; Reyes, G.; Knoflach, F.; Crestani, F.; Aguzzi, A.; Arigoni, M.; Lang, Y.; et al. Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. USA 1995, 92, 7749–7753. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Q. The GABAA Receptor 2 Subunit R43Q Mutation Linked to Childhood Absence Epilepsy and Febrile Seizures Causes Retention of 1 2 2S Receptors in the Endoplasmic Reticulum. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 8672–8677. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hernandez, C.C.; Hu, N.; Macdonald, R.L. Three epilepsy-associated GABRG2 missense mutations at the γ+/β− interface disrupt GABAA receptor assembly and trafficking by similar mechanisms but to different extents. Neurobiol. Dis. 2014, 68, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Q.; Shen, W.; Zhou, C.; Xu, D.; Macdonald, R.L. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat. Neurosci. 2015, 18, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Q.; Shen, W.; Lee, M.; Gallagher, M.J.; Macdonald, R.L. Slow Degradation and Aggregation In Vitro of Mutant GABAA Receptor 2(Q351X) Subunits Associated with Epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 13895–13905. [Google Scholar] [CrossRef]
- Kang, J.-Q.; Shen, W.; Macdonald, R.L. The GABRG2 Mutation, Q351X, Associated with Generalized Epilepsy with Febrile Seizures Plus, Has Both Loss of Function and Dominant-Negative Suppression. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2845–2856. [Google Scholar] [CrossRef]
- Warner, T.A.; Shen, W.; Huang, X.; Liu, Z.; Macdonald, R.L.; Kang, J.-Q. Differential molecular and behavioural alterations in mouse models of GABRG2haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum. Mol. Genet. 2016, 25, 3192–3207. [Google Scholar] [CrossRef]
- Tanaka, M.; DeLorey, T.M.; Delgado-Escueta, A.; Olsen, R.W. GABRB3, Epilepsy, and Neurodevelopment. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK98178/ (accessed on 23 March 2021).
- Carvill, G.L.; Weckhuysen, S.; McMahon, J.M.; Hartmann, C.; Møller, R.S.; Hjalgrim, H.; Cook, J.; Geraghty, E.; O’Roak, B.J.; Petrou, S.; et al. GABRA1 and STXBP1: Novel genetic causes of Dravet syndrome. Neurology 2014, 82, 1245–1253. [Google Scholar] [CrossRef]
- Le, S.V.; Le, P.H.T.; Van Le, T.K.; Huynh, T.T.K.; Do, T.T.H. A mutation in GABRB3 associated with Dravet syndrome. Am. J. Med Genet. Part A 2017, 173, 2126–2131. [Google Scholar] [CrossRef]
- Vithlani, M.; Moss, S.J. The role of GABAAR phosphorylation in the construction of inhibitory synapses and the efficacy of neuronal inhibition. Biochem. Soc. Trans. 2009, 37, 1355–1358. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.-M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De Novo Mutations in Moderate or Severe Intellectual Disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef] [PubMed]
- Urak, L.; Feucht, M.; Fathi, N.; Hornik, K.; Fuchs, K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum. Mol. Genet. 2006, 15, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Epi4K Consortium, Epilepsy Phenome/Genome Project; Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Adak, P.; Sinha, S.; Banerjee, N. An Association Study of Gamma-Aminobutyric Acid Type A Receptor Variants and Susceptibility to Autism Spectrum Disorders. J. Autism Dev. Disord. 2021, 51, 4043–4053. [Google Scholar] [CrossRef] [PubMed]
- Feucht, M.; Fuchs, K.; Pichlbauer, E.; Hornik, K.; Scharfetter, J.; Goessler, R.; Füreder, T.; Cvetkovic, N.; Sieghart, W.; Kasper, S.; et al. Possible association between childhood absence epilepsy and the gene encoding GABRB3. Biol. Psychiatry 1999, 46, 997–1002. [Google Scholar] [CrossRef]
- Ms, V.S.J.; Hernandez, C.C.; Bs, K.M.V.; Hu, N.; Macdonald, R.L. Epileptic encephalopathy de novoGABRBmutations impair γ-aminobutyric acid type A receptor function. Ann. Neurol. 2016, 79, 806–825. [Google Scholar] [CrossRef]
- Shi, Y.-W.; Zhang, Q.; Cai, K.; Poliquin, S.; Shen, W.; Winters, N.; Yi, Y.-H.; Wang, J.; Hu, N.; Macdonald, R.L.; et al. Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 2019, 142, 3028–3044. [Google Scholar] [CrossRef]
- Chen, W.; Ge, Y.; Lu, J.; Melo, J.; So, Y.W.; Juneja, R.; Liu, L.; Wang, Y.T. Distinct Functional Alterations and Therapeutic Options of Two Pathological De Novo Variants of the T292 Residue of GABRA1 Identified in Children with Epileptic Encephalopathy and Neurodevelopmental Disorders. Int. J. Mol. Sci. 2022, 23, 2723. [Google Scholar] [CrossRef]
- Demarest, S.T.; Olson, H.E.; Moss, A.; Pestana-Knight, E.; Zhang, X.; Parikh, S.; Swanson, L.C.; Riley, K.D.; Bazin, G.A.; Angione, K.; et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia 2019, 60, 1733–1742. [Google Scholar] [CrossRef]
- Mantegazza, M.; Gambardella, A.; Rusconi, R.; Schiavon, E.; Annesi, F.; Cassulini, R.R.; Labate, A.; Carrideo, S.; Chifari, R.; Canevini, M.P.; et al. Identification of an Nav 1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl. Acad. Sci. USA 2005, 102, 18177–18182. [Google Scholar] [CrossRef] [Green Version]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.F.; Wagnon, J.L.; Mefford, H.C.; Meisler, M.H. SCN8A-Related Epilepsy with Encephalopathy. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., et al., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Goto, A.; Ishii, A.; Shibata, M.; Ihara, Y.; Cooper, E.C.; Hirose, S. Characteristics of KCNQ 2 variants causing either benign neonatal epilepsy or developmental and epileptic encephalopathy. Epilepsia 2019, 60, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Singhal, N.; El Achkar, C.M.; Truglio, G.; Sheidley, B.R.; Sullivan, J.; Poduri, A. PCDH19 -related epilepsy is associated with a broad neurodevelopmental spectrum. Epilepsia 2018, 59, 679–689. [Google Scholar] [CrossRef]
- Tye, C.; Thomas, L.; Sampson, J.R.; Lewis, J.; O’Callaghan, F.; Yates, J.R.W.; Bolton, P.F. Secular changes in severity of intellectual disability in tuberous sclerosis complex: A reflection of improved identification and treatment of epileptic spasms? Epilepsia Open 2018, 3, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef]
- Cai, K.; Wang, J.; Eissman, J.; Wang, J.; Nwosu, G.; Shen, W.; Liang, H.-C.; Li, X.-J.; Zhu, H.-X.; Yi, Y.-H.; et al. A missense mutation in SLC6A1 associated with Lennox-Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp. Neurol. 2019, 320, 112973. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; van Roon-Mom, W.M.; Marckmann, G.; van Duyvenvoorde, H.A.; Graessner, H.; Schüle, R.; Aartsma-Rus, A. Preparing n-of-1 Antisense Oligonucleotide Treatments for Rare Neurological Diseases in Europe: Genetic, Regulatory, and Ethical Perspectives. Nucleic Acid Ther. 2022, 32, 83–94. [Google Scholar] [CrossRef]
- Mirza, N.; Stevelink, R.; Taweel, B.; Koeleman, B.P.C.; Marson, A.G.; Abou-Khalil, B.; Auce, P.; Avbersek, A.; Bahlo, M.; Balding, D.J.; et al. Using common genetic variants to find drugs for common epilepsies. Brain Commun. 2021, 3, fcab287. [Google Scholar] [CrossRef]
- Symonds, J.; Zuberi, S.M.; Johnson, M.R. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr. Opin. Neurol. 2017, 30, 193–199. [Google Scholar] [CrossRef]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic en-cephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Thakran, S.; Guin, D.; Singh, P.; Singh, P.; Kukal, S.; Rawat, C.; Yadav, S.; Kushwaha, S.S.; Srivastava, A.K.; Hasija, Y.; et al. Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment. Int. J. Mol. Sci. 2020, 21, 7784. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nwosu, G.; Reddy, S.B.; Riordan, H.R.M.; Kang, J.-Q. Variable Expression of GABAA Receptor Subunit Gamma 2 Mutation in a Nuclear Family Displaying Developmental and Encephalopathic Phenotype. Int. J. Mol. Sci. 2022, 23, 9683. https://doi.org/10.3390/ijms23179683
Nwosu G, Reddy SB, Riordan HRM, Kang J-Q. Variable Expression of GABAA Receptor Subunit Gamma 2 Mutation in a Nuclear Family Displaying Developmental and Encephalopathic Phenotype. International Journal of Molecular Sciences. 2022; 23(17):9683. https://doi.org/10.3390/ijms23179683
Chicago/Turabian StyleNwosu, Gerald, Shilpa B. Reddy, Heather Rose Mead Riordan, and Jing-Qiong Kang. 2022. "Variable Expression of GABAA Receptor Subunit Gamma 2 Mutation in a Nuclear Family Displaying Developmental and Encephalopathic Phenotype" International Journal of Molecular Sciences 23, no. 17: 9683. https://doi.org/10.3390/ijms23179683
APA StyleNwosu, G., Reddy, S. B., Riordan, H. R. M., & Kang, J. -Q. (2022). Variable Expression of GABAA Receptor Subunit Gamma 2 Mutation in a Nuclear Family Displaying Developmental and Encephalopathic Phenotype. International Journal of Molecular Sciences, 23(17), 9683. https://doi.org/10.3390/ijms23179683