

Clinically Relevant KCNQ1 Variants Causing KCNQ1-KCNE2 Gain-of-Function Affect the Ca2+ Sensitivity of the Channel

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Identification of a Previously Reported and Novel KCNQ1 Pathogenic Variant in Individuals with Gingival Overgrowth
2.2. Analysis of the Novel KCNQ1 Variant V185M Reveals Gain-of-Function
2.3. Mutant KCNQ1 Channels Exhibit Reduced Current Run-Down
2.4. High Intracellular Ca2+ and CaM Counteract Early Current Run-Down
2.5. Mutant KCNQ1 Channels Exhibit Impaired Ca2+ Sensitivity
2.6. Impaired Ca2+ Sensitivity of KCNQ1 Mutant Channels Causes Gain-of-Function of Heteromeric KCNQ1-KCNE2 Channels
2.7. Corroboration of the Crucial Role of Ca2+ in Q1E2 Channel Activation in Somato-Mammotroph Anterior Pituitary Cells
3. Discussion
4. Materials and Methods
4.1. Whole-Exome Sequencing, Variant Calling and Segregation Analysis
4.2. Site-Directed Mutagenesis
4.3. Cell Culture
4.4. Heterologous Expression
4.5. Solutions and Special Experimental Conditions
4.6. Electrophysiology
4.7. Data Analysis
4.8. Statistics
4.9. Model Generation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individual | 1 | 2 | 3 (Son of Individual 2) | 4 (Sister of Individual 2) |
|---|---|---|---|---|
| Gene | KCNQ1 | KCNQ1 | KCNQ1 | KCNQ1 |
| mRNA reference number | NM_000218.3 | NM_000218.3 | NM_000218.3 | NM_000218.3 |
| Variant | c.1106C>T p.Pro369Leu | c.553G>A p.Val185Met | c.553G>A p.Val185Met | c.553G>A p.Val185Met |
| Worldwide MAF of the variant (gnomAD 2.1.1) | Absent | 0.00001 | 0.00001 | 0.00001 |
| Origin | Unknown | unknown | Maternally inherited | unknown |
| Nationality | Caucasian (Egyptian) | Caucasian (Portuguese) | Caucasian (Portuguese) | Caucasian (Portuguese) |
| Sex | Female | Female | Male | Female |
| Age at last examination | 5 years and 9 months | 38 years | 16 years and 11 months | 38 years |
| Birth weight | 3500 g (full term) (0.57 z) | ND | ND | ND |
| Birth length | 48 cm (−0.6 z) | ND | ND | ND |
| OFC at birth | ND | ND | ND | ND |
| Weight at last examination | 16 kg (−1.91 z) | 68.2 kg BMI: 26.2 | 55 kg (−1.53 z) | 90 kg BMI: 35.2 |
| Height at last examination | 102 cm (−2.82 z) | 161 cm (−1.1 z) | 165 cm (−2 z) | 160 cm (−1.26 z) |
| OFC at last examination | 51 cm (0.05 z) | 54 cm (−1.03 z) | 55.5 cm (−0.77 z) | ND |
| Craniofacial dysmorphism | Coarse face, upslanted palpebral fissures, bulbous nose, thickened ears with fleshy lobes, wide open mouth, high-arched and narrow palate, failure of eruption of primary teeth | Long face, broad nasal tip, full lips, retrognathia | Long face, broad nasal tip, full lips | Coarse face, thick eyebrows, retrognathia, high-arched palate |
| Gingival overgrowth | + (progressive since early infancy) | + (since childhood) | + (since birth) | + (since infancy) |
| Pituitary hormone deficiencies | T3, T4, and thyroid-stimulating hormone normal, IGF-1 very low, GH deficiency (maximum GH after clonidine and L dopa: 0.76 ng/mL; normal response >10) | ND | ND | ND |
| Developmental delay | − | − | + | Learning difficulties |
| MRI scan | Cavum septum pellucidum and cavum vergae, a small simple choroid plexus cyst seen at the trigone of the right lateral ventricle, otherwise normal | ND | ND | ND |
| Cardiovascularfeatures | Normal echocardiogram and electrocardiogram | Normal echocardio-gram and electrocardio-gram | ND | Normal electrocardiogram |
| Other anomalies | Long fingers, hypertrichosis on back, abnormality of the voice | − | Long fingers | Brachydactyly |
Appendix B


References
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Seebohm, G. Physiological Functions, Biophysical Properties, and Regulation of KCNQ1 (KV7.1) Potassium Channels. Adv. Exp. Med. Biol. 2021, 1349, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Pusch, M.; Magrassi, R.; Wollnik, B.; Conti, F. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 1998, 75, 785–792. [Google Scholar] [CrossRef]
- Ghosh, S.; Nunziato, D.A.; Pitt, G.S. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 2006, 98, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Shamgar, L.; Ma, L.; Schmitt, N.; Haitin, Y.; Peretz, A.; Wiener, R.; Hirsch, J.; Pongs, O.; Attali, B. Calmodulin is essential for cardiac IKS channel gating and assembly: Impaired function in long-QT mutations. Circ. Res. 2006, 98, 1055–1063. [Google Scholar] [CrossRef]
- Li, Y.; Zaydman, M.A.; Wu, D.; Shi, J.; Guan, M.; Virgin-Downey, B.; Cui, J. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc. Natl. Acad. Sci. USA 2011, 108, 9095–9100. [Google Scholar] [CrossRef]
- Li, Y.; Gao, J.; Lu, Z.; McFarland, K.; Shi, J.; Bock, K.; Cohen, I.S.; Cui, J. Intracellular ATP binding is required to activate the slowly activating K+ channel IKs. Proc. Natl. Acad. Sci. USA 2013, 110, 18922–18927. [Google Scholar] [CrossRef]
- Zaydman, M.A.; Silva, J.R.; Delaloye, K.; Li, Y.; Liang, H.; Larsson, H.P.; Shi, J.; Cui, J. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. USA 2013, 110, 13180–13185. [Google Scholar] [CrossRef]
- Tobelaim, W.S.; Dvir, M.; Lebel, G.; Cui, M.; Buki, T.; Peretz, A.; Marom, M.; Haitin, Y.; Logothetis, D.E.; Hirsch, J.A.; et al. Competition of calcified calmodulin N lobe and PIP2 to an LQT mutation site in Kv7.1 channel. Proc. Natl. Acad. Sci. USA 2017, 114, E869–E878. [Google Scholar] [CrossRef]
- Yus-Najera, E.; Santana-Castro, I.; Villarroel, A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 2002, 277, 28545–28553. [Google Scholar] [CrossRef] [Green Version]
- Sachyani, D.; Dvir, M.; Strulovich, R.; Tria, G.; Tobelaim, W.; Peretz, A.; Pongs, O.; Svergun, D.; Attali, B.; Hirsch, J.A. Structural basis of a Kv7.1 potassium channel gating module: Studies of the intracellular C-terminal domain in complex with calmodulin. Structure 2014, 22, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Tobelaim, W.S.; Dvir, M.; Lebel, G.; Cui, M.; Buki, T.; Peretz, A.; Marom, M.; Haitin, Y.; Logothetis, D.E.; Hirsch, J.A.; et al. Ca2+-Calmodulin and PIP2 interactions at the proximal C-terminus of Kv7 channels. Channels 2017, 11, 686–695. [Google Scholar] [CrossRef] [PubMed]
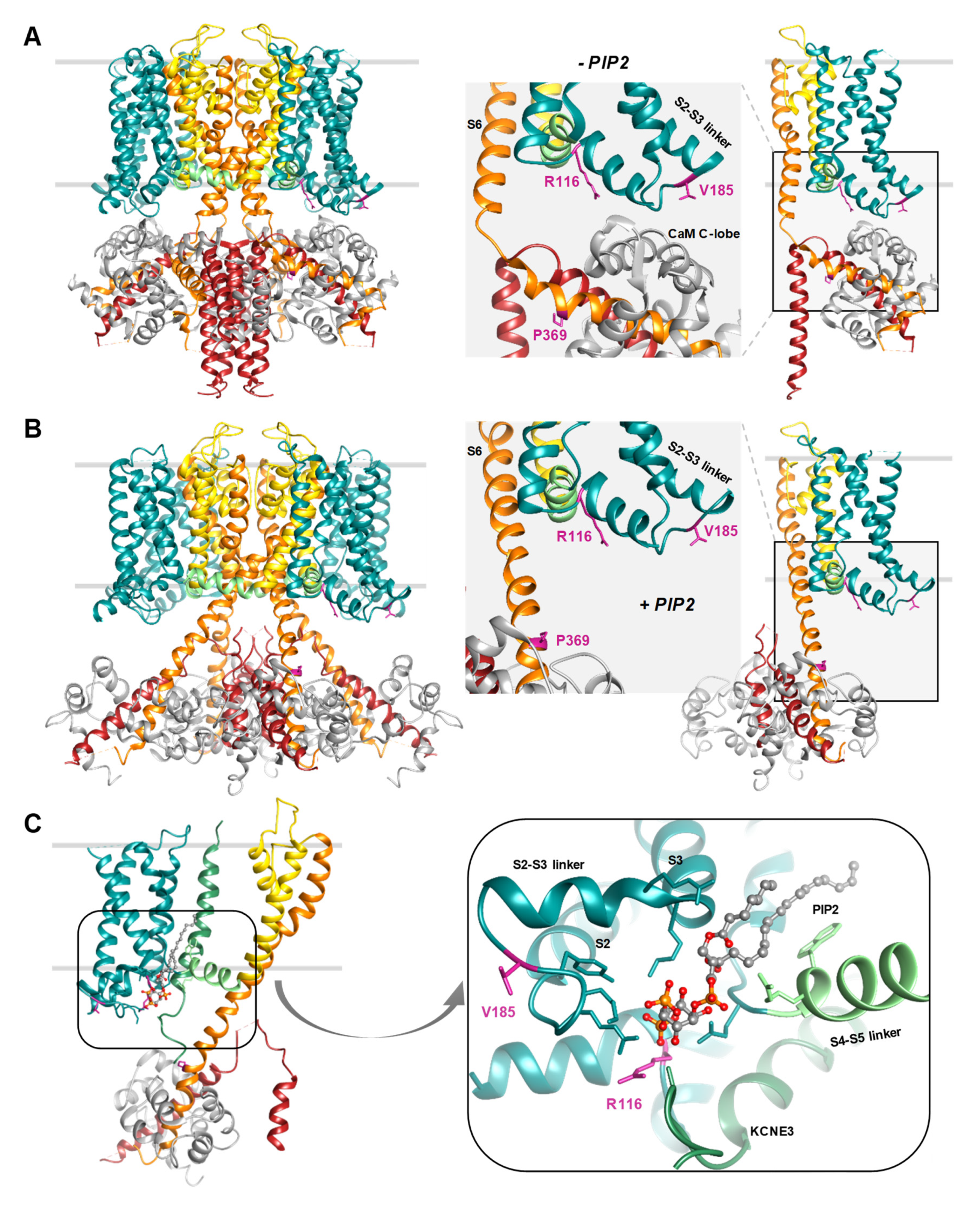
- Sun, J.; MacKinnon, R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 2017, 169, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347. [Google Scholar] [CrossRef]
- Kang, P.W.; Westerlund, A.M.; Shi, J.; White, K.M.; Dou, A.K.; Cui, A.H.; Silva, J.R.; Delemotte, L.; Cui, J. Calmodulin acts as a state-dependent switch to control a cardiac potassium channel opening. Sci. Adv. 2020, 6, eabd6798. [Google Scholar] [CrossRef]
- Abbott, G.W. Biology of the KCNQ1 Potassium Channel. New J. Sci. 2014, 2014, 237431. [Google Scholar] [CrossRef]
- Jones, F.; Gamper, N.; Gao, H. Kv7 Channels and Excitability Disorders. In Pharmacology of Potassium Channels; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2021; Volume 267, pp. 185–230. [Google Scholar] [CrossRef]
- Wang, Y.; Eldstrom, J.; Fedida, D. Gating and Regulation of KCNQ1 and KCNQ1 + KCNE1 Channel Complexes. Front. Physiol. 2020, 11, 504. [Google Scholar] [CrossRef]
- Pongs, O.; Schwarz, J.R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 2010, 90, 755–796. [Google Scholar] [CrossRef]
- Tommiska, J.; Kansakoski, J.; Skibsbye, L.; Vaaralahti, K.; Liu, X.; Lodge, E.J.; Tang, C.; Yuan, L.; Fagerholm, R.; Kanters, J.K.; et al. Two missense mutations in KCNQ1 cause pituitary hormone deficiency and maternally inherited gingival fibromatosis. Nat. Commun. 2017, 8, 1289. [Google Scholar] [CrossRef] [Green Version]
- Tinel, N.; Diochot, S.; Borsotto, M.; Lazdunski, M.; Barhanin, J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 2000, 19, 6326–6330. [Google Scholar] [CrossRef]
- Heitzmann, D.; Grahammer, F.; von Hahn, T.; Schmitt-Graff, A.; Romeo, E.; Nitschke, R.; Gerlach, U.; Lang, H.J.; Verrey, F.; Barhanin, J.; et al. Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J. Physiol. 2004, 561, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W. The KCNE2 K+ channel regulatory subunit: Ubiquitous influence, complex pathobiology. Gene 2015, 569, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.; Eldstrom, J.; Fedida, D. Hormonal Signaling Actions on Kv7.1 (KCNQ1) Channels. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Stojilkovic, S.S.; Tabak, J.; Bertram, R. Ion channels and signaling in the pituitary gland. Endocr. Rev. 2010, 31, 845–915. [Google Scholar] [CrossRef] [PubMed]
- Simons, C.; Rash, L.D.; Crawford, J.; Ma, L.; Cristofori-Armstrong, B.; Miller, D.; Ru, K.; Baillie, G.J.; Alanay, Y.; Jacquinet, A.; et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat. Genet. 2015, 47, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Kortum, F.; Caputo, V.; Bauer, C.K.; Stella, L.; Ciolfi, A.; Alawi, M.; Bocchinfuso, G.; Flex, E.; Paolacci, S.; Dentici, M.L.; et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 2015, 47, 661–667. [Google Scholar] [CrossRef]
- Bauer, C.K.; Calligari, P.; Radio, F.C.; Caputo, V.; Dentici, M.L.; Falah, N.; High, F.; Pantaleoni, F.; Barresi, S.; Ciolfi, A.; et al. Mutations in KCNK4 that Affect Gating Cause a Recognizable Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2018, 103, 621–630. [Google Scholar] [CrossRef]
- Bauer, C.K.; Schneeberger, P.E.; Kortum, F.; Altmuller, J.; Santos-Simarro, F.; Baker, L.; Keller-Ramey, J.; White, S.M.; Campeau, P.M.; Gripp, K.W.; et al. Gain-of-Function Mutations in KCNN3 Encoding the Small-Conductance Ca2+-Activated K+ Channel SK3 Cause Zimmermann-Laband Syndrome. Am. J. Hum. Genet. 2019, 104, 1139–1157. [Google Scholar] [CrossRef]
- Gripp, K.W.; Smithson, S.F.; Scurr, I.J.; Baptista, J.; Majumdar, A.; Pierre, G.; Williams, M.; Henderson, L.B.; Wentzensen, I.M.; McLaughlin, H.; et al. Syndromic disorders caused by gain-of-function variants in KCNH1, KCNK4, and KCNN3-a subgroup of K+ channelopathies. Eur. J. Hum. Genet. 2021, 29, 1384–1395. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Suri, M. “Electrifying dysmorphology”: Potassium channelopathies causing dysmorphic syndromes. Adv. Genet. 2020, 105, 137–174. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, C.; Meng, L.; Wang, Z.; Chen, D.; Peng, Y.; Yang, K.; Bian, Z. Activated KCNQ1 channel promotes fibrogenic response in hereditary gingival fibromatosis via clustering and activation of Ras. J. Periodontal Res. 2021, 56, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. KVLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, T.; Rasmussen, H.B.; Grunnet, M.; Jensen, H.S.; Angelo, K.; Dupuis, D.S.; Vogel, L.K.; Jorgensen, N.K.; Klaerke, D.A.; Olesen, S.P. Basolateral localisation of KCNQ1 potassium channels in MDCK cells: Molecular identification of an N-terminal targeting motif. J. Cell Sci. 2004, 117, 4517–4526. [Google Scholar] [CrossRef]
- Melman, Y.F.; Domenech, A.; de la Luna, S.; McDonald, T.V. Structural determinants of KvLQT1 control by the KCNE family of proteins. J. Biol. Chem. 2001, 276, 6439–6444. [Google Scholar] [CrossRef]
- Loussouarn, G.; Park, K.H.; Bellocq, C.; Baro, I.; Charpentier, F.; Escande, D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: A functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003, 22, 5412–5421. [Google Scholar] [CrossRef]
- Wulfsen, I.; Hauber, H.P.; Schiemann, D.; Bauer, C.K.; Schwarz, J.R. Expression of mRNA for voltage-dependent and inward-rectifying K channels in GH3/B6 cells and rat pituitary. J. Neuroendocrinol. 2000, 12, 263–272. [Google Scholar] [CrossRef]
- Schledermann, W.; Wulfsen, I.; Schwarz, J.R.; Bauer, C.K. Modulation of rat erg1, erg2, erg3 and HERG K+ currents by thyrotropin-releasing hormone in anterior pituitary cells via the native signal cascade. J. Physiol. 2001, 532, 143–163. [Google Scholar] [CrossRef]
- Borgini, M.; Mondal, P.; Liu, R.; Wipf, P. Chemical modulation of Kv7 potassium channels. RSC Med. Chem. 2021, 12, 483–537. [Google Scholar] [CrossRef]
- Abbott, G.W.; Redford, K.E.; Yoshimura, R.F.; Manville, R.W.; Moreira, L.; Tran, K.; Arena, G.; Kookootsedes, A.; Lasky, E.; Gunnison, E. KCNQ and KCNE Isoform-Dependent Pharmacology Rationalizes Native American Dual Use of Specific Plants as Both Analgesics and Gastrointestinal Therapeutics. Front. Physiol. 2021, 12, 777057. [Google Scholar] [CrossRef]
- Tristani-Firouzi, M.; Sanguinetti, M.C. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 1998, 510, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Shi, J.; White, K.M.; Gao, Y.; Cui, J. ML277 specifically enhances the fully activated open state of KCNQ1 by modulating VSD-pore coupling. eLife 2019, 8, e48576. [Google Scholar] [CrossRef]
- Klemic, K.G.; Shieh, C.C.; Kirsch, G.E.; Jones, S.W. Inactivation of Kv2.1 potassium channels. Biophys. J. 1998, 74, 1779–1789. [Google Scholar] [CrossRef]
- Zaydman, M.A.; Kasimova, M.A.; McFarland, K.; Beller, Z.; Hou, P.; Kinser, H.E.; Liang, H.; Zhang, G.; Shi, J.; Tarek, M.; et al. Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. eLife 2014, 3, e03606. [Google Scholar] [CrossRef] [PubMed]
- Tzounopoulos, T.; Maylie, J.; Adelman, J.P. Gating of IsK channels expressed in Xenopus oocytes. Biophys. J. 1998, 74, 2299–2305. [Google Scholar] [CrossRef]
- Strutz-Seebohm, N.; Pusch, M.; Wolf, S.; Stoll, R.; Tapken, D.; Gerwert, K.; Attali, B.; Seebohm, G. Structural basis of slow activation gating in the cardiac IKs channel complex. Cell. Physiol. Biochem. 2011, 27, 443–452. [Google Scholar] [CrossRef]
- Chang, A.; Abderemane-Ali, F.; Hura, G.L.; Rossen, N.D.; Gate, R.E.; Minor, D.L., Jr. A Calmodulin C-Lobe Ca2+-Dependent Switch Governs Kv7 Channel Function. Neuron 2018, 97, 836–852.e6. [Google Scholar] [CrossRef]
- Gamper, N.; Li, Y.; Shapiro, M.S. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol. Biol. Cell 2005, 16, 3538–3551. [Google Scholar] [CrossRef]
- Lerche, C.; Scherer, C.R.; Seebohm, G.; Derst, C.; Wei, A.D.; Busch, A.E.; Steinmeyer, K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 2000, 275, 22395–22400. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Q.; Qiu, Y.; Li, Z.; Chen, Z.; Jiang, H.; Li, Y.; Yang, H. Migration of PIP2 lipids on voltage-gated potassium channel surface influences channel deactivation. Sci. Rep. 2015, 5, 15079. [Google Scholar] [CrossRef]
- Zhuang, W.; Yan, Z. The S2-S3 Loop of Kv7.4 Channels Is Essential for Calmodulin Regulation of Channel Activation. Front. Physiol. 2021, 11, 604134. [Google Scholar] [CrossRef] [PubMed]
- Lehman, A.; Thouta, S.; Mancini, G.M.S.; Naidu, S.; van Slegtenhorst, M.; McWalter, K.; Person, R.; Mwenifumbo, J.; Salvarinova, R.; Study, C.; et al. Loss-of-Function and Gain-of-Function Mutations in KCNQ5 Cause Intellectual Disability or Epileptic Encephalopathy. Am. J. Hum. Genet. 2017, 101, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.D.; Wakenight, P.; Zwingman, T.A.; Bard, A.M.; Sahai, N.; Willemsen, M.H.; Schelhaas, H.J.; Stegmann, A.P.A.; Verhoeven, J.S.; de Man, S.A.; et al. Human KCNQ5 de novo Mutations Underlie Epilepsy and Intellectual Disability. J. Neurophysiol. 2022, 128, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Nappi, M.; Barrese, V.; Carotenuto, L.; Lesca, G.; Labalme, A.; Ville, D.; Smol, T.; Rama, M.; Dieux-Coeslier, A.; Rivier-Ringenbach, C.; et al. Gain of function due to increased opening probability by two KCNQ5 pore variants causing developmental and epileptic encephalopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2116887119. [Google Scholar] [CrossRef] [PubMed]
- Dahimene, S.; Alcolea, S.; Naud, P.; Jourdon, P.; Escande, D.; Brasseur, R.; Thomas, A.; Baro, I.; Merot, J. The N-terminal juxtamembranous domain of KCNQ1 is critical for channel surface expression: Implications in the Romano-Ward LQT1 syndrome. Circ. Res. 2006, 99, 1076–1083. [Google Scholar] [CrossRef]
- Roepke, T.K.; King, E.C.; Reyna-Neyra, A.; Paroder, M.; Purtell, K.; Koba, W.; Fine, E.; Lerner, D.J.; Carrasco, N.; Abbott, G.W. Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat. Med. 2009, 15, 1186–1194. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Wiel, L.; Baakman, C.; Gilissen, D.; Veltman, J.A.; Vriend, G.; Gilissen, C. MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat 2019, 40, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Altman, R.B.; Bromberg, Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genom. 2013, 14 (Suppl. 3), S2. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.S.; Strehler, E.E. The tumor-sensitive calmodulin-like protein is a specific light chain of human unconventional myosin X. J. Biol. Chem. 2001, 276, 12182–12189. [Google Scholar] [CrossRef]
- Tang, Q.; Jin, M.W.; Xiang, J.Z.; Dong, M.Q.; Sun, H.Y.; Lau, C.P.; Li, G.R. The membrane permeable calcium chelator BAPTA-AM directly blocks human ether a-go-go-related gene potassium channels stably expressed in HEK 293 cells. Biochem. Pharmacol. 2007, 74, 1596–1607. [Google Scholar] [CrossRef]
- Watkins, C.S.; Mathie, A. Effects on K+ currents in rat cerebellar granule neurones of a membrane-permeable analogue of the calcium chelator BAPTA. Br. J. Pharmacol. 1996, 118, 1772–1778. [Google Scholar] [CrossRef] [Green Version]
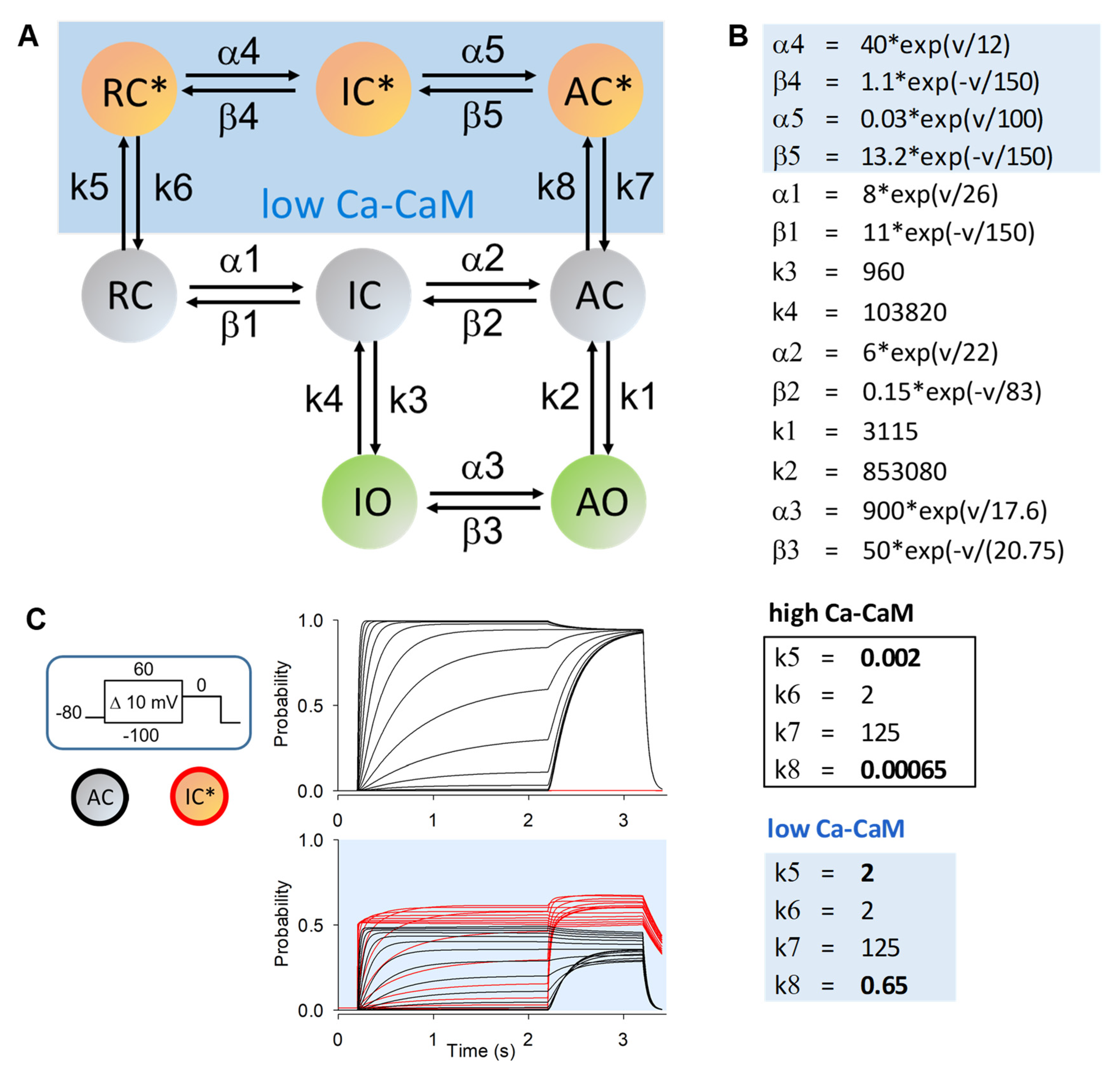
- Zifarelli, G.; Zuccolini, P.; Bertelli, S.; Pusch, M. The Joy of Markov Models—Channel Gating and Transport Cycling Made Easy. Biophysicist 2021, 2, 70–107. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, C.K.; Holling, T.; Horn, D.; Laço, M.N.; Abdalla, E.; Omar, O.M.; Alawi, M.; Kutsche, K. Clinically Relevant KCNQ1 Variants Causing KCNQ1-KCNE2 Gain-of-Function Affect the Ca2+ Sensitivity of the Channel. Int. J. Mol. Sci. 2022, 23, 9690. https://doi.org/10.3390/ijms23179690
Bauer CK, Holling T, Horn D, Laço MN, Abdalla E, Omar OM, Alawi M, Kutsche K. Clinically Relevant KCNQ1 Variants Causing KCNQ1-KCNE2 Gain-of-Function Affect the Ca2+ Sensitivity of the Channel. International Journal of Molecular Sciences. 2022; 23(17):9690. https://doi.org/10.3390/ijms23179690
Chicago/Turabian StyleBauer, Christiane K., Tess Holling, Denise Horn, Mário Nôro Laço, Ebtesam Abdalla, Omneya Magdy Omar, Malik Alawi, and Kerstin Kutsche. 2022. "Clinically Relevant KCNQ1 Variants Causing KCNQ1-KCNE2 Gain-of-Function Affect the Ca2+ Sensitivity of the Channel" International Journal of Molecular Sciences 23, no. 17: 9690. https://doi.org/10.3390/ijms23179690
APA StyleBauer, C. K., Holling, T., Horn, D., Laço, M. N., Abdalla, E., Omar, O. M., Alawi, M., & Kutsche, K. (2022). Clinically Relevant KCNQ1 Variants Causing KCNQ1-KCNE2 Gain-of-Function Affect the Ca2+ Sensitivity of the Channel. International Journal of Molecular Sciences, 23(17), 9690. https://doi.org/10.3390/ijms23179690