
Integrated Analysis of Transcriptome and Metabolome Reveals the Regulation of Chitooligosaccharide on Drought Tolerance in Sugarcane (Saccharum spp. Hybrid) under Drought Stress

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect Analysis of Roc22 Phenotype and Physiology under Different Treatments
2.2. Transcriptome Analysis
2.3. Metabolome Analysis
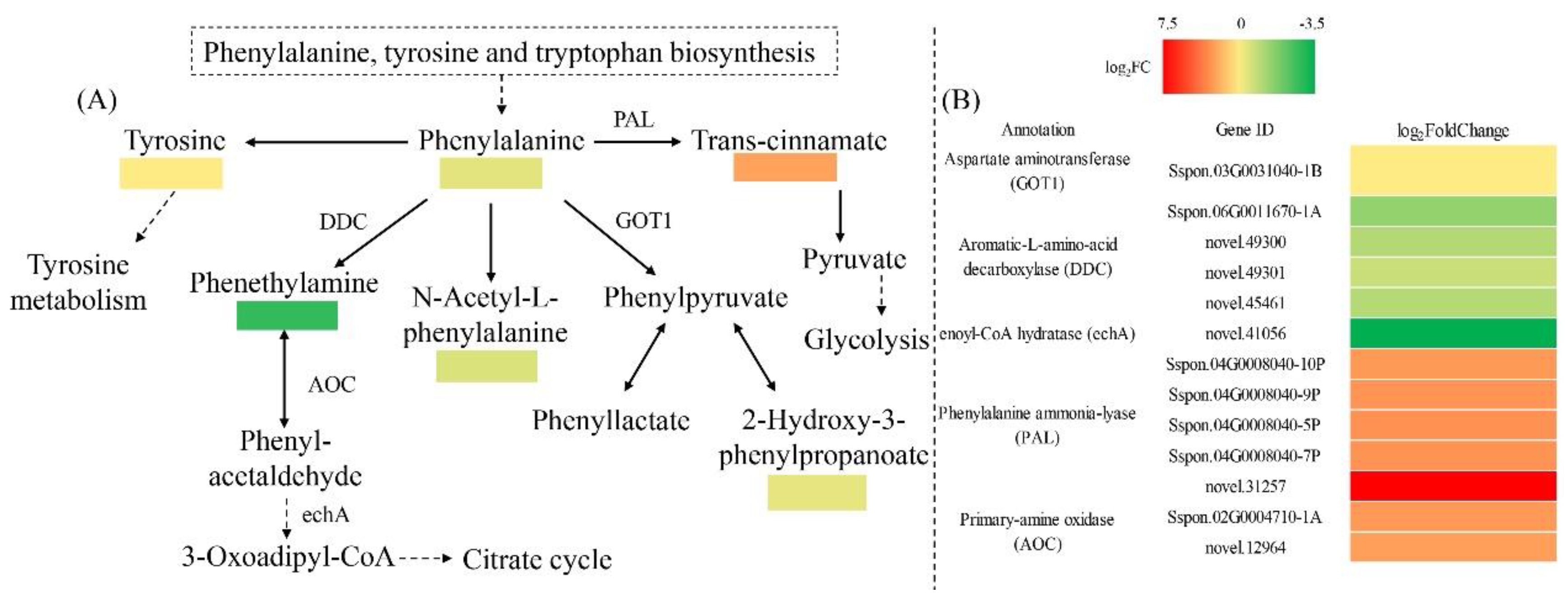
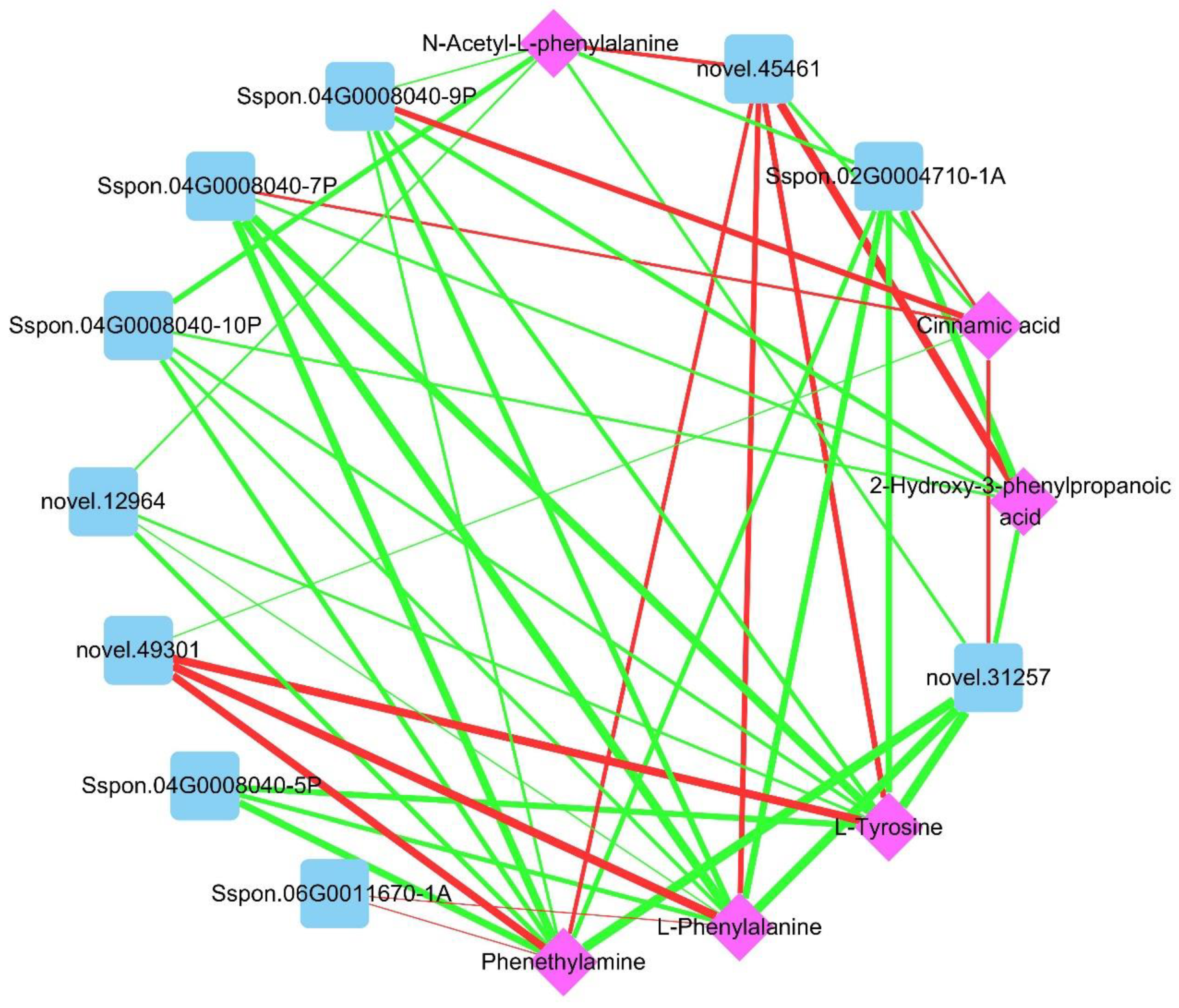
2.4. Combined Transcriptome and Metabolome Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Drought Stress
4.2. Measurement of Physiological Parameters
4.3. Rna Sequencing and Data Analysis
4.4. Real-Time Quantitative Pcr (Rt-qpcr)
4.5. Widely Targeted Metabolomics and Data Analysis
4.6. UPLC Conditions
4.7. ESI-Q TRAP-MS/MS
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, A.; Khan, M.; Sharif, R.; Mujtaba, M.; Gao, S.J. Sugarcane omics: An update on the current status of research and crop improvement. Plants 2019, 8, 344. [Google Scholar] [CrossRef]
- Fàbregas, N.; Fernie, A.R. The metabolic response to drought. J. Exp. Bot. 2019, 70, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lu, M.; Wang, Y.; Wang, Y.; Liu, Z.; Chen, S. Response mechanism of plants to drought stress. Horticulturae 2021, 7, 50. [Google Scholar] [CrossRef]
- Leanasawat, N.; Kosittrakun, M.; Lontom, W.; Songsri, P. Physiological and agronomic traits of certain sugarcane genotypes grown under field conditions as influenced by early drought stress. Agronomy 2021, 11, 2319. [Google Scholar] [CrossRef]
- Vital, C.E.; Giordano, A.; de Almeida Soares, E.; Rhys Williams, T.C.; Mesquita, R.O.; Vidigal, P.M.P.; de Santana Lopes, A.; Pacheco, T.G.; Rogalski, M.; de Oliveira Ramos, H.J.; et al. An integrative overview of the molecular and physiological responses of sugarcane under drought conditions. Plant Mol. Biol. 2017, 94, 577–594. [Google Scholar] [CrossRef] [PubMed]
- McMillan, T.; Tidemann, B.D.; O’Donovan, J.T.; Izydorczyk, M.S. Effects of plant growth regulator application on the malting quality of barley. J. Sci. Food Agr. 2020, 100, 2082–2089. [Google Scholar] [CrossRef]
- Rostami, S.; Azhdarpoor, A. The application of plant growth regulators to improve phytoremediation of contaminated soils: A review. Chemosphere 2019, 220, 818–827. [Google Scholar] [CrossRef]
- Bielach, A.; Duclercq, J.; Marhavý, P.; Benková, E. Genetic approach towards the identification of auxin-cytokinin crosstalk components involved in root development. Phil. Trans. R. Soc. B 2012, 367, 1469–1478. [Google Scholar] [CrossRef]
- He, Y.; Zhao, J.; Yang, B.; Sun, S.; Peng, L.; Wang, Z. Indole-3-acetate beta-glucosyltransferase OsIAGLU regulates seed vigour through mediating crosstalk between auxin and abscisic acid in rice. Plant Biotechnol. J. 2020, 18, 1933–1945. [Google Scholar] [CrossRef]
- Yuan, T.T.; Xu, H.H.; Zhang, K.X.; Guo, T.T.; Lu, Y.T. Glucose inhibits root meristem growth via ABA INSENSITIVE 5, which represses PIN1 accumulation and auxin activity in Arabidopsis. Plant Cell Environ. 2014, 37, 1338–1350. [Google Scholar] [CrossRef]
- Kocięcka, J.; Liberacki, D. The potential of using chitosan on cereal crops in the face of climate change. Plants 2021, 10, 1160. [Google Scholar] [CrossRef] [PubMed]
- Muley, A.B.; Shingote, P.R.; Patil, A.P.; Dalvi, S.G.; Suprasanna, P. Gamma radiation degradation of chitosan for application in growth promotion and induction of stress tolerance in potato (Solanum tuberosum L.). Carbohyd. Polym. 2019, 210, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chai, Z.; Lin, P.; Huang, C.; Huang, G.; Xu, L.; Deng, Z.; Zhang, M.; Zhang, Y.; Zhao, X. Genome-wide identification and expression analysis of AP2/ERF transcription factors in sugarcane (Saccharum spontaneum L.). BMC Genom. 2020, 21, 685. [Google Scholar] [CrossRef] [PubMed]
- Selvi, A.; Devi, K.; Manimekalai, R.; Prathima, P.T. Comparative analysis of drought-responsive transcriptomes of sugarcane genotypes with differential tolerance to drought. 3 Biotech 2020, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, R.; Saito, K. Integrated metabolomics for abiotic stress responses in plants. Curr. Opin. Plant Biol. 2015, 24, 10–16. [Google Scholar] [CrossRef]
- Vijayaraghavareddy, P.; Akula, N.N.; Vemanna, R.S.; Math, R.G.H.; Shinde, D.D.; Yin, X.; Struik, P.C.; Makarla, U.; Sreeman, S. Metabolome profiling reveals impact of water limitation on grain filling in contrasting rice genotypes. Plant Physiol. Bioch. 2021, 162, 690–698. [Google Scholar] [CrossRef]
- Ferreira, D.A.; Martins, M.C.M.; Cheavegatti-Gianotto, A.; Carneiro, M.S.; Amadeu, R.R.; Aricetti, J.A.; Wolf, L.D.; Hoffmann, H.P.; de Abreu, L.G.F.; Caldana, C. Metabolite profiles of sugarcane culm reveal the relationship among metabolism and axillary bud outgrowth in genetically related sugarcane commercial cultivars. Front. Plant. Sci. 2018, 9, 857. [Google Scholar] [CrossRef]
- Ni, Y.; Chen, H.; Liu, D.; Zeng, L.; Chen, P.; Liu, C. Discovery of genes involved in anthocyanin biosynthesis from the rind and pith of three sugarcane varieties using integrated metabolic profiling and RNA-seq analysis. BMC Plant Biol. 2021, 21, 214. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.; Gong, Z.; Zhu, J.K. Abiotic stress responses in plants. Nat. Rev. Genet. 2022, 23, 104–119. [Google Scholar] [CrossRef]
- Dinh, H.T.; Watanable, K.; Takaragawa, H.; Kawamitsu, Y. Effects of Drought Stress at Early Growth Stage on Response of Sugarcane to Different Nitrogen Application. Sugar Tech 2018, 20, 420–430. [Google Scholar] [CrossRef]
- Endres, L.; dos Santos, C.M.; Silva, J.V.; Barbosa, G.V.D.; Silva, A.L.J.; Froehlich, A.; Teixeira, M.M. Inter-relationship between photosynthetic efficiency, delta C-13, antioxidant activity and sugarcane yield under drought stress in field conditions. J. Agron. Crop Sci. 2019, 205, 433–446. [Google Scholar] [CrossRef]
- Abbas, S.R.; Ahmad, S.D.; Sabir, S.M.; Shah, A.H. Detection of drought tolerant sugarcane genotypes (Saccharum officinarum) using lipid peroxidation, antioxidant activity, glycine-betaine and proline contents. J. Soil Plant Nut. 2014, 14, 233–243. [Google Scholar] [CrossRef]
- Moolphuerk, N.; Pattanagul, W. Pretreatment with different molecular weight chitosans encourages drought tolerance in rice (Oryza sativa L.) seedling. Not. Bot. Horti Agrobo. 2020, 48, 2072–2084. [Google Scholar] [CrossRef]
- Hafez, Y.; Attia, K.; Alamery, S.; Ghazy, A.; Al-Doss, A.; Ibrahim, E.; Rashwan, E.; El-Maghraby, L.; Awad, A.; Abdelaal, K. Beneficial effects of biochar and chitosan on antioxidative capacity, osmolytes accumulation, and anatomical characters of water-stressed barley plants. Agronomy 2020, 10, 630. [Google Scholar] [CrossRef]
- Herppich, W.B.; Peckmann, K. Influence of drought on mitochondrial activity, photosynthesis, nocturnal acid accumulation and water relations in the CAM Plants Prenia sladeniana (ME-type) and Crassula lycopodioides (PEPCK-type). Ann. Bot.-Lond. 2000, 86, 611–620. [Google Scholar] [CrossRef]
- Pagter, M.; Bragato, C.; Brix, H. Tolerance and physiological responses of Phragmites australis to water deficit. Aquat. Bot. 2005, 81, 285–299. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, W.; Zhang, F.; Deng, J.; Li, Z.; Fu, B. Comparative metabolite profiling of two rice genotypes with contrasting salt stress tolerance at the seedling stage. PLoS ONE 2014, 9, e108020. [Google Scholar] [CrossRef]
- Yi, D.; Zhang, H.; Lai, B.; Liu, L.; Pan, X.; Ma, Z.; Wang, Y.; Xie, J.; Shi, S.; Wei, Y. Integrative analysis of the coloring mechanism of red longan pericarp through metabolome and transcriptome analyses. J. Agric. Food Chem. 2021, 69, 1806–1815. [Google Scholar] [CrossRef]
- Le, X.H.; Lee, C.P.; Millar, A.H. The mitochondrial pyruvate carrier (MPC) complex mediates one of three pyruvate-supplying pathways that sustain Arabidopsis respiratory metabolism. Plant Cell 2021, 33, 2776–2793. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Park, C.J.; Ham, B.K.; Choi, S.B.; Lee, B.J.; Paek, K.H. Induction of a cytosolic pyruvate kinase 1 gene during the resistance response to tobacco mosaic virus in Capsicum annuum. Plant Cell Rep. 2006, 25, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, W.; Luo, L.; Pang, J.; Rong, W.; He, C. Downregulation of OsPK1, a cytosolic pyruvate kinase, by T-DNA insertion causes dwarfism and panicle enclosure in rice. Planta 2012, 235, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Li, S.; Jiao, G.; Sheng, Z.; Wu, Y.; Shao, G.; Xie, L.; Peng, C.; Xu, J.; Tang, S.; et al. OsPK2 encodes a plastidic pyruvate kinase involved in rice endosperm starch synthesis, compound granule formation and grain filling. Plant Biotechnol. J. 2018, 16, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, W.; Jin, J.; Yang, X.; You, X.; Yan, H.; Wang, L.; Chen, J.; Xu, J.; Chen, W.; et al. OsPKpα1 encodes a plastidic pyruvate kinase that affects starch biosynthesis in the rice endosperm. J. Integ. Plant Biol. 2018, 60, 1097–1118. [Google Scholar] [CrossRef]
- Frelin, O.; Dervinis, C.; Wegrzyn, J.L.; Davis, J.M.; Hanson, A.D. Drought stress in Pinus taeda L. induces coordinated transcript accumulation of genes involved in the homogentisate pathway. Tree Genet. Genomes 2017, 13, 27. [Google Scholar] [CrossRef]
- Feng, Y.T.; Huang, Q.L.; Zhang, R.; Li, J.Y.; Luo, K.; Chen, Y.H. Molecular characterisation of PAL gene family reveals their role in abiotic stress response in lucerne (Medicago sativa). Crop Pasture Sci. 2022, 73, 300–311. [Google Scholar] [CrossRef]
- Mo, F.Y.; Li, L.; Zhang, C.; Yang, C.H.; Chen, G.; Niu, Y.; Si, J.X.; Liu, T.; Sun, X.X.; Wang, S.L.; et al. Genome-wide analysis and expression profiling of the phenylalanine ammonia-lyase gene family in Solanum tuberosum. Int. J. Mol. Sci. 2022, 23, 6833. [Google Scholar] [CrossRef]
- Sova, M. Antioxidant and antimicrobial activities of cinnamic acid derivatives. Mini-Rev. Med. Chem. 2012, 12, 749–767. [Google Scholar] [CrossRef]
- Yin, X.; Liu, S.; Qin, Y.; Xing, R.; Li, K.; Yu, C.; Chen, X.; Li, P. Metabonomics analysis of drought resistance of wheat seedlings induced by β-aminobutyric acid-modified chitooligosaccharide derivative. Carbohyd. Polym. 2021, 272, 118437. [Google Scholar] [CrossRef]
- Moustakas, M.; Sperdouli, I.; Kouna, T.; Antonopoulou, C.I.; Therios, I. Exogenous proline induces soluble sugar accumulation and alleviates drought stress effects on photosystem II functioning of Arabidopsis thaliana leaves. Plant Growth Regul. 2011, 65, 315–325. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Determination of malondialdehyde as dithiobarbituric acid adduct in biological samples by HPLC with fluorescence detection: Comparison with ultraviolet-visible spectrophotometry. Clin. Chem. 2001, 47, 1725–1727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Chu, N.; Zhou, H.; Li, J.; Feng, N.; Su, J.; Deng, Z.; Shen, X.; Zheng, D. Integrated Analysis of Transcriptome and Metabolome Reveals the Regulation of Chitooligosaccharide on Drought Tolerance in Sugarcane (Saccharum spp. Hybrid) under Drought Stress. Int. J. Mol. Sci. 2022, 23, 9737. https://doi.org/10.3390/ijms23179737
Yang S, Chu N, Zhou H, Li J, Feng N, Su J, Deng Z, Shen X, Zheng D. Integrated Analysis of Transcriptome and Metabolome Reveals the Regulation of Chitooligosaccharide on Drought Tolerance in Sugarcane (Saccharum spp. Hybrid) under Drought Stress. International Journal of Molecular Sciences. 2022; 23(17):9737. https://doi.org/10.3390/ijms23179737
Chicago/Turabian StyleYang, Shan, Na Chu, Hongkai Zhou, Jiashuo Li, Naijie Feng, Junbo Su, Zuhu Deng, Xuefeng Shen, and Dianfeng Zheng. 2022. "Integrated Analysis of Transcriptome and Metabolome Reveals the Regulation of Chitooligosaccharide on Drought Tolerance in Sugarcane (Saccharum spp. Hybrid) under Drought Stress" International Journal of Molecular Sciences 23, no. 17: 9737. https://doi.org/10.3390/ijms23179737
APA StyleYang, S., Chu, N., Zhou, H., Li, J., Feng, N., Su, J., Deng, Z., Shen, X., & Zheng, D. (2022). Integrated Analysis of Transcriptome and Metabolome Reveals the Regulation of Chitooligosaccharide on Drought Tolerance in Sugarcane (Saccharum spp. Hybrid) under Drought Stress. International Journal of Molecular Sciences, 23(17), 9737. https://doi.org/10.3390/ijms23179737