
The Oncolytic Adenovirus XVir-N-31, in Combination with the Blockade of the PD-1/PD-L1 Axis, Conveys Abscopal Effects in a Humanized Glioblastoma Mouse Model


Abstract
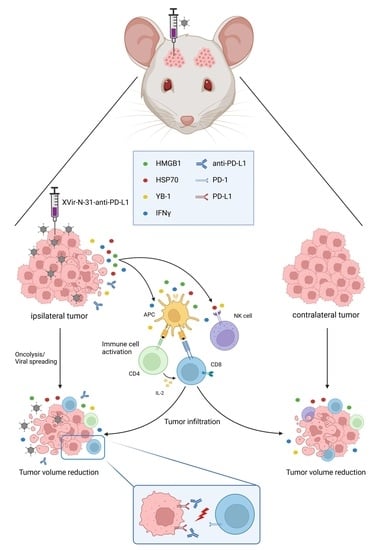
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Determination of Virus Mediated Cell Killing Activity and Virus Functionality
2.2. XVir-N-31 and XVir-N-31-Anti-PD-L1 Induce Immunogenic Cell Death
2.3. Virus Mediated IFNγ Release
2.4. XVir-N-31 Leads to an Elevated Number of TILs That Can Be Further Raised by Immune Checkpoint Inhibition
2.5. XVir-N-31-Anti-PD-L1 or XVir-N-31 in Combination with Nivolumab Presented an Abscopal Effect in the Reduction in Tumor Growth
3. Discussion
4. Material and Methods
4.1. Cells, Cell Lines, and Cell Culture
4.2. Adenoviral Vectors and Infection
4.3. Cell Viability Assays
4.4. Determination of Virus Copy Numbers
4.5. Immunoblot Analyses
4.6. PD-1/PD-L1 Binding Assay
4.7. ELISA Based Detection of HMGB1, HSP70, YB-1 and IFNγ
4.8. Flow Cytometry Analysis (FACS)
4.9. Immuno-Humanized Mouse Model
4.10. Histology and Immunofluorescence
4.11. Hematoxylin and Eosin (H&E) Staining and Tumor Volumetry
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ad-WT | Wildtype adenovirus (subtype 5) |
| APC | Allophycocyanin |
| CTLA4 | Cytotoxic T-lymphocyte associated protein 4 |
| CNS | Central nervous system |
| CRT | Calreticulin |
| CTL | Cytotoxic T lymphocyte |
| DAMP | Danger associated molecular pattern |
| DAPI | 4′,6-Diamidino-2-phenylindol |
| DC | Dendritic cell |
| DMEM | Dulbecco’s modified Eagle’s medium |
| EDTA | Ethylendiamintetraacetate |
| FACS | Fluorescence Activated Cell Sorting |
| FCS | Fetal calf serum |
| FITC | Fluorescein isothiocyanate |
| GAPDH | Glycerinaldehyd-3-phosphat-Dehydrogenase |
| GBM | Glioblastoma |
| HA | Hemaglutinin |
| HLA | Human leukocyte antigene |
| ICD | Immunogenic cell death |
| HMGB1 | High mobility group protein 1 |
| HSP70 | Heat shock protein 70 |
| ICI | Immune checkpoint inhibition |
| ICP | Immune checkpoint protein |
| IFNγ | Interferon gamma |
| IL | Interleukin |
| MOI | Moiety of infection |
| MTT | 3-(4,5-Methylthiazol)-2,5-diphenyltetrazolium bromide |
| NK | Natural killer |
| OAV | Oncolytic adenovirus |
| OV | Oncolytic virus |
| OVT | Oncolytic virotherapy |
| PAMP | Pathogen associated molecular pattern |
| PBMC | Peripheral blood mononuclear cells |
| PBS | Phosphate buffered saline |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death ligand 1 |
| p.i. | Post infection |
| P/S | Penicillin/streptomycin |
| TILs | Tumor infiltrating lymphocytes |
| Treg | Regulatory T cell |
| UKT | University Hospital Tübingen |
| YB-1 | Y-box protein 1 |
References
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. Jama 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Zeyaullah, M.; Patro, M.; Ahmad, I.; Ibraheem, K.; Sultan, P.; Nehal, M.; Ali, A. Oncolytic viruses in the treatment of cancer: A review of current strategies. Pathol. Oncol. Res. 2012, 18, 771–781. [Google Scholar] [CrossRef]
- Kosnopfel, C.; Sinnberg, T.; Schittek, B. Y-box binding protein 1—A prognostic marker and target in tumour therapy. Eur. J. Cell Biol. 2014, 93, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Jing, W.; Orentas, R.J. Discovery of YB-1 as a new immunological target in neuroblastoma by vaccination in the context of regulatory T cell blockade. Acta Biochim. Biophys. Sin. 2009, 41, 980–990. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e1110. [Google Scholar] [CrossRef]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharmacol. 2017, 8, 242. [Google Scholar] [CrossRef] [PubMed]
- Akhbariyoon, H.; Azizpour, Y.; Esfahani, M.F.; Firoozabad, M.S.M.; Rad, M.R.; Esfahani, K.S.; Khoshavi, N.; Karimi, N.; Shirinisaz, A.; Abedi, F.; et al. Immune checkpoint inhibition for the treatment of cancers: An update and critical review of ongoing clinical trials. Clin. Immunol. 2021, 232, 108873. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q. Immune checkpoint inhibitors in GBM. J. Neurooncol. 2021, 155, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, H.; Chen, B.; Liu, X.; Zhang, S.; Zong, Z.; Gao, M. PD-L1-Mediated Immunosuppression in Glioblastoma Is Associated With the Infiltration and M2-Polarization of Tumor-Associated Macrophages. Front. Immunol. 2020, 11, 588552. [Google Scholar] [CrossRef]
- Pan, P.C.; Haggiagi, A. Neurologic Immune-Related Adverse Events Associated with Immune Checkpoint Inhibition. Curr. Oncol. Rep. 2019, 21, 108. [Google Scholar] [CrossRef]
- Rognoni, E.; Widmaier, M.; Haczek, C.; Mantwill, K.; Holzmuller, R.; Gansbacher, B.; Kolk, A.; Schuster, T.; Schmid, R.M.; Saur, D.; et al. Adenovirus-based virotherapy enabled by cellular YB-1 expression in vitro and in vivo. Cancer Gene Ther. 2009, 16, 753–763. [Google Scholar] [CrossRef]
- Czolk, R.; Schwarz, N.; Koch, H.; Schotterl, S.; Wuttke, T.V.; Holm, P.S.; Huber, S.M.; Naumann, U. Irradiation enhances the therapeutic effect of the oncolytic adenovirus XVir-N-31 in brain tumor initiating cells. Int. J. Mol. Med. 2019, 44, 1484–1494. [Google Scholar] [CrossRef]
- Hindupur, S.V.; Schmid, S.C.; Koch, J.A.; Youssef, A.; Baur, E.M.; Wang, D.; Horn, T.; Slotta-Huspenina, J.; Gschwend, J.E.; Holm, P.S.; et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 1106. [Google Scholar] [CrossRef]
- Lichtenegger, E.; Koll, F.; Haas, H.; Mantwill, K.; Janssen, K.P.; Laschinger, M.; Gschwend, J.; Steiger, K.; Black, P.C.; Moskalev, I.; et al. The Oncolytic Adenovirus XVir-N-31 as a Novel Therapy in Muscle-Invasive Bladder Cancer. Hum. Gene Ther. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Wang, J.Z.; Zhu, H.; You, P.; Liu, H.; Wang, W.K.; Fan, X.; Yang, Y.; Xu, K.; Zhu, Y.; Li, Q.; et al. Upregulated YB-1 protein promotes glioblastoma growth through a YB-1/CCT4/mLST8/mTOR pathway. J. Clin. Investig. 2022, 132, e146536. [Google Scholar] [CrossRef]
- Alayo, Q.A.; Ito, H.; Passaro, C.; Zdioruk, M.; Mahmoud, A.B.; Grauwet, K.; Zhang, X.; Lawler, S.E.; Reardon, D.A.; Goins, W.F.; et al. Glioblastoma infiltration of both tumor- and virus-antigen specific cytotoxic T cells correlates with experimental virotherapy responses. Sci. Rep. 2020, 10, 5095. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Rabkin, S.D. Immunohistochemistry for Tumor-Infiltrating Immune Cells After Oncolytic Virotherapy. Methods Mol. Biol. 2020, 2058, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Passaro, C.; Alayo, Q.; De Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-chain Fragment Variable Antibody against PD-1 for Experimental Glioblastoma Therapy. Clin. Cancer Res. 2019, 25, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2017, 19, 493–502. [Google Scholar] [CrossRef]
- Puigdelloses, M.; Garcia-Moure, M.; Labiano, S.; Laspidea, V.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; De la Nava, D.; Ausejo, I.; et al. CD137 and PD-L1 targeting with immunovirotherapy induces a potent and durable antitumor immune response in glioblastoma models. J. Immunother. Cancer 2021, 9, e002644. [Google Scholar] [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020, 11, 48. [Google Scholar] [CrossRef]
- Mantwill, K.; Naumann, U.; Seznec, J.; Girbinger, V.; Lage, H.; Surowiak, P.; Beier, D.; Mittelbronn, M.; Schlegel, J.; Holm, P.S. YB-1 dependent oncolytic adenovirus efficiently inhibits tumor growth of glioma cancer stem like cells. J. Transl. Med. 2013, 11, 216. [Google Scholar] [CrossRef]
- Oliveira, E.R.A.; Bouvier, M. Immune evasion by adenoviruses: A window into host-virus adaptation. FEBS Lett. 2019, 593, 3496–3503. [Google Scholar] [CrossRef]
- Anghelina, D.; Lam, E.; Falck-Pedersen, E. Diminished Innate Antiviral Response to Adenovirus Vectors in cGAS/STING-Deficient Mice Minimally Impacts Adaptive Immunity. J. Virol. 2016, 90, 5915–5927. [Google Scholar] [CrossRef]
- Iribarren, K.; Buque, A.; Mondragon, L.; Xie, W.; Levesque, S.; Pol, J.; Zitvogel, L.; Kepp, O.; Kroemer, G. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology 2019, 8, 1550619. [Google Scholar] [CrossRef] [Green Version]
- Ylosmaki, E.; Ylosmaki, L.; Fusciello, M.; Martins, B.; Ahokas, P.; Cojoc, H.; Uoti, A.; Feola, S.; Kreutzman, A.; Ranki, T.; et al. Characterization of a novel OX40 ligand and CD40 ligand-expressing oncolytic adenovirus used in the PeptiCRAd cancer vaccine platform. Mol. Ther. Oncolytics 2021, 20, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Pellegrini, M.; Horwitz, G.A.; Xie, W.; Berk, A.J.; Kurdistani, S.K. Epigenetic reprogramming by adenovirus e1a. Science 2008, 321, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Tone, Y.; Kojima, Y.; Furuuchi, K.; Brady, M.; Yashiro-Ohtani, Y.; Tykocinski, M.L.; Tone, M. OX40 gene expression is up-regulated by chromatin remodeling in its promoter region containing Sp1/Sp3, YY1, and NF-kappa B binding sites. J. Immunol. 2007, 179, 1760–1767. [Google Scholar] [CrossRef]
- Pelka, P.; Ablack, J.N.; Shuen, M.; Yousef, A.F.; Rasti, M.; Grand, R.J.; Turnell, A.S.; Mymryk, J.S. Identification of a second independent binding site for the pCAF acetyltransferase in adenovirus E1A. Virology 2009, 391, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, G.A.; Zhang, K.; McBrian, M.A.; Grunstein, M.; Kurdistani, S.K.; Berk, A.J. Adenovirus small e1a alters global patterns of histone modification. Science 2008, 321, 1084–1085. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.R.; Grigera, F.; Ucker, D.S.; Cook, J.L. Adenovirus E1B 19-kilodalton protein modulates innate immunity through apoptotic mimicry. J. Virol. 2014, 88, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, J.A.; Mertens, P.R. Cold shock proteins: From cellular mechanisms to pathophysiology and disease. Cell Commun. Signal. 2018, 16, 63. [Google Scholar] [CrossRef]
- Kitamura, N.; Murata, S.; Ueki, T.; Mekata, E.; Reilly, R.T.; Jaffee, E.M.; Tani, T. OX40 costimulation can abrogate Foxp3+ regulatory T cell-mediated suppression of antitumor immunity. Int. J. Cancer 2009, 125, 630–638. [Google Scholar] [CrossRef]
- Workenhe, S.T.; Simmons, G.; Pol, J.G.; Lichty, B.D.; Halford, W.P.; Mossman, K.L. Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol. Ther. 2014, 22, 123–131. [Google Scholar] [CrossRef]
- Kakiuchi, Y.; Kuroda, S.; Kanaya, N.; Kumon, K.; Tsumura, T.; Hashimoto, M.; Yagi, C.; Sugimoto, R.; Hamada, Y.; Kikuchi, S.; et al. Local oncolytic adenovirotherapy produces an abscopal effect via tumor-derived extracellular vesicles. Mol. Ther. 2021, 29, 2920–2930. [Google Scholar] [CrossRef]
- Jaime-Sanchez, P.; Uranga-Murillo, I.; Aguilo, N.; Khouili, S.C.; Arias, M.A.; Sancho, D.; Pardo, J. Cell death induced by cytotoxic CD8(+) T cells is immunogenic and primes caspase-3-dependent spread immunity against endogenous tumor antigens. J. Immunother. Cancer 2020, 8, e000528. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11, 582106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, C.; Liu, X.; Sun, L.; Li, G.; Liang, J.; Hu, H.; Liu, Y.; Zhang, W.; Jiang, T. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology 2016, 5, e1196310. [Google Scholar] [CrossRef]
- Hamdan, F.; Ylosmaki, E.; Chiaro, J.; Giannoula, Y.; Long, M.; Fusciello, M.; Feola, S.; Martins, B.; Feodoroff, M.; Antignani, G.; et al. Novel oncolytic adenovirus expressing enhanced cross-hybrid IgGA Fc PD-L1 inhibitor activates multiple immune effector populations leading to enhanced tumor killing in vitro, in vivo and with patient-derived tumor organoids. J. Immunother. Cancer 2021, 9, e003000. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Ramachandran, M.; Lugano, R.; Ma, J.; Martikainen, M.M.; Dimberg, A.; Yu, D.; Merits, A.; Essand, M. IFN-I-tolerant oncolytic Semliki Forest virus in combination with anti-PD1 enhances T cell response against mouse glioma. Mol. Ther. Oncolytics 2021, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Mazzoccoli, L.; Jash, A.; Govero, J.; Bais, S.S.; Hu, T.; Fontes-Garfias, C.R.; Shan, C.; Okada, H.; Shresta, S.; et al. Zika virus oncolytic activity requires CD8+ T cells and is boosted by immune checkpoint blockade. JCI Insight 2021, 6, e144619. [Google Scholar] [CrossRef] [PubMed]
- Panagioti, E.; Kurokawa, C.; Viker, K.; Ammayappan, A.; Anderson, S.K.; Sotiriou, S.; Chatzopoulos, K.; Ayasoufi, K.; Johnson, A.J.; Iankov, I.D.; et al. Immunostimulatory bacterial antigen-armed oncolytic measles virotherapy significantly increases the potency of anti-PD1 checkpoint therapy. J. Clin. Investig. 2021, 131, 141614. [Google Scholar] [CrossRef]
- Vijayakumar, G.; McCroskery, S.; Palese, P. Engineering Newcastle Disease Virus as an Oncolytic Vector for Intratumoral Delivery of Immune Checkpoint Inhibitors and Immunocytokines. J. Virol. 2020, 94, e01677-19. [Google Scholar] [CrossRef]
- Hibma, M.H.; Real, N.C.; Wiles, A.; Dobson-Le, D.; Dix, B.R.; Wynford-Thomas, D.; Braithwaite, A.W.; Royds, J.A. Increased apoptosis and reduced replication efficiency of the E3 region-modified dl309 adenovirus in cancer cells. Virus Res. 2009, 145, 112–120. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “humanized” mice to study human immunity. Curr. Protoc. Immunol. 2008, 81, 15.21.1–15.21.21. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Humanized SCID mouse models for biomedical research. Curr. Top Microbiol. Immunol. 2008, 324, 25–51. [Google Scholar] [PubMed]
- Ehx, G.; Somja, J.; Warnatz, H.J.; Ritacco, C.; Hannon, M.; Delens, L.; Fransolet, G.; Delvenne, P.; Muller, J.; Beguin, Y.; et al. Xenogeneic Graft-Versus-Host Disease in Humanized NSG and NSG-HLA-A2/HHD Mice. Front. Immunol. 2018, 9, 1943. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.; Isenmann, S.; Naumann, U.; Kugler, S.; Bahr, M.; Dichgans, J.; Ashkenazi, A.; Weller, M. Locoregional Apo2L/TRAIL eradicates intracranial human malignant glioma xenografts in athymic mice in the absence of neurotoxicity. Biochem. Biophys. Res. Commun. 1999, 265, 479–483. [Google Scholar] [CrossRef]
- Armento, A.; Ilina, E.I.; Kaoma, T.; Muller, A.; Vallar, L.; Niclou, S.P.; Kruger, M.A.; Mittelbronn, M.; Naumann, U. Carboxypeptidase E transmits its anti-migratory function in glioma cells via transcriptional regulation of cell architecture and motility regulating factors. Int. J. Oncol. 2017, 51, 702–714. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klawitter, M.; El-Ayoubi, A.; Buch, J.; Rüttinger, J.; Ehrenfeld, M.; Lichtenegger, E.; Krüger, M.A.; Mantwill, K.; Koll, F.J.; Kowarik, M.C.; et al. The Oncolytic Adenovirus XVir-N-31, in Combination with the Blockade of the PD-1/PD-L1 Axis, Conveys Abscopal Effects in a Humanized Glioblastoma Mouse Model. Int. J. Mol. Sci. 2022, 23, 9965. https://doi.org/10.3390/ijms23179965
Klawitter M, El-Ayoubi A, Buch J, Rüttinger J, Ehrenfeld M, Lichtenegger E, Krüger MA, Mantwill K, Koll FJ, Kowarik MC, et al. The Oncolytic Adenovirus XVir-N-31, in Combination with the Blockade of the PD-1/PD-L1 Axis, Conveys Abscopal Effects in a Humanized Glioblastoma Mouse Model. International Journal of Molecular Sciences. 2022; 23(17):9965. https://doi.org/10.3390/ijms23179965
Chicago/Turabian StyleKlawitter, Moritz, Ali El-Ayoubi, Jasmin Buch, Jakob Rüttinger, Maximilian Ehrenfeld, Eva Lichtenegger, Marcel A. Krüger, Klaus Mantwill, Florestan J. Koll, Markus C. Kowarik, and et al. 2022. "The Oncolytic Adenovirus XVir-N-31, in Combination with the Blockade of the PD-1/PD-L1 Axis, Conveys Abscopal Effects in a Humanized Glioblastoma Mouse Model" International Journal of Molecular Sciences 23, no. 17: 9965. https://doi.org/10.3390/ijms23179965
APA StyleKlawitter, M., El-Ayoubi, A., Buch, J., Rüttinger, J., Ehrenfeld, M., Lichtenegger, E., Krüger, M. A., Mantwill, K., Koll, F. J., Kowarik, M. C., Holm, P. S., & Naumann, U. (2022). The Oncolytic Adenovirus XVir-N-31, in Combination with the Blockade of the PD-1/PD-L1 Axis, Conveys Abscopal Effects in a Humanized Glioblastoma Mouse Model. International Journal of Molecular Sciences, 23(17), 9965. https://doi.org/10.3390/ijms23179965