
Unusual Cytochrome c552 from Thioalkalivibrio paradoxus: Solution NMR Structure and Interaction with Thiocyanate Dehydrogenase

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. CytC552 Is Characteristic of the Genus Thioalkalivibrio Strains Expressing TcDH and Serves as Its Electron Acceptor
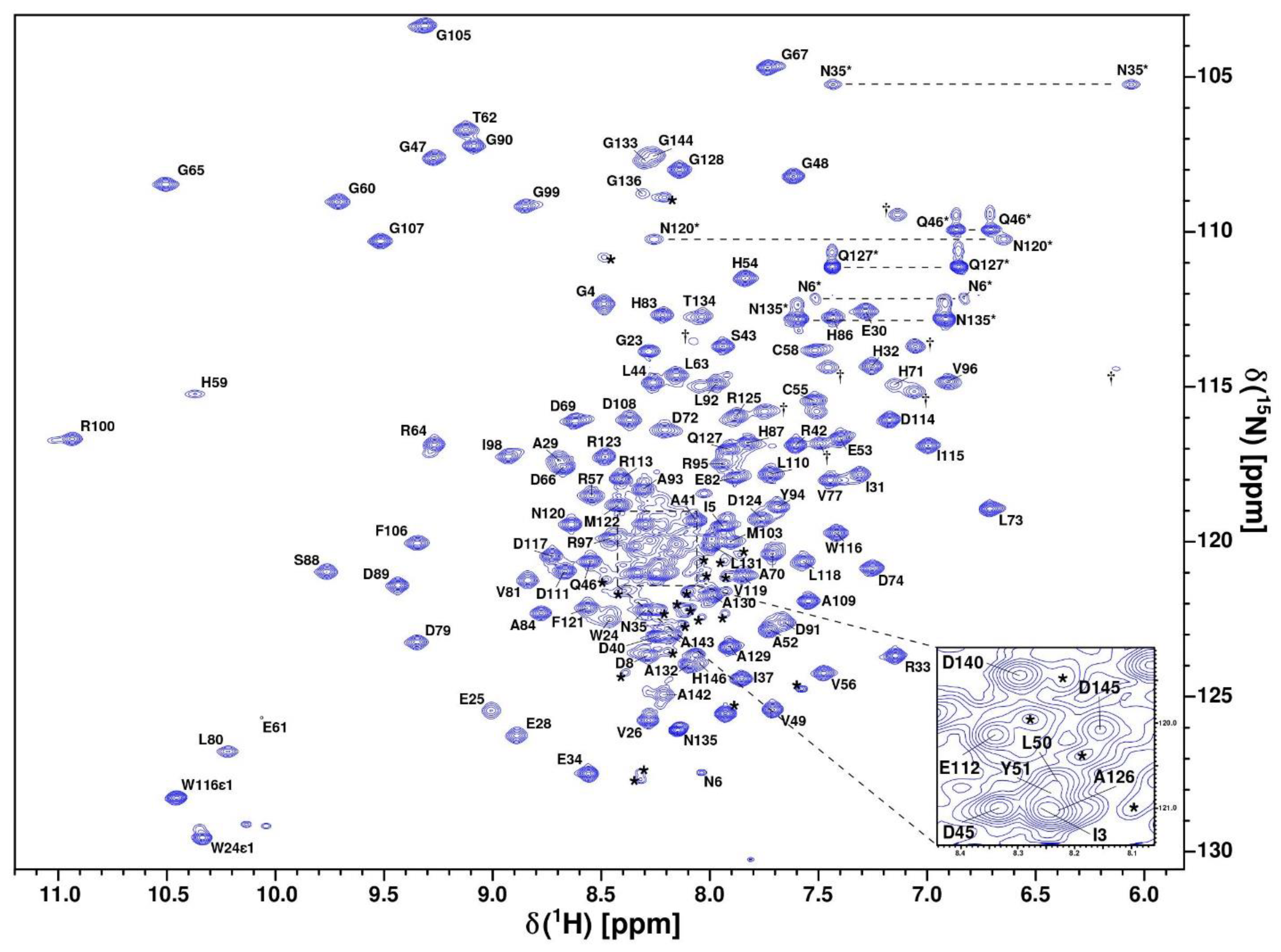
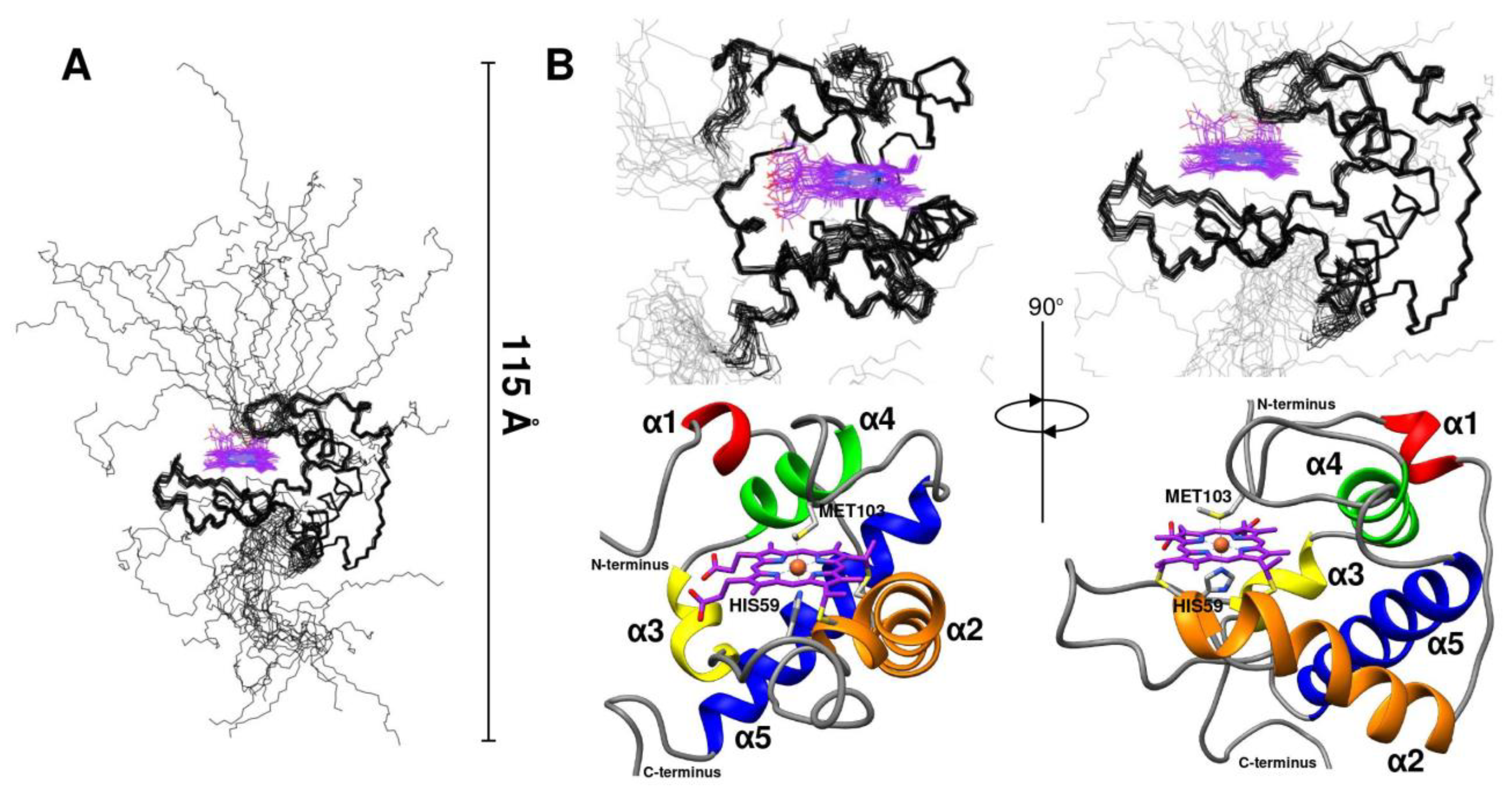
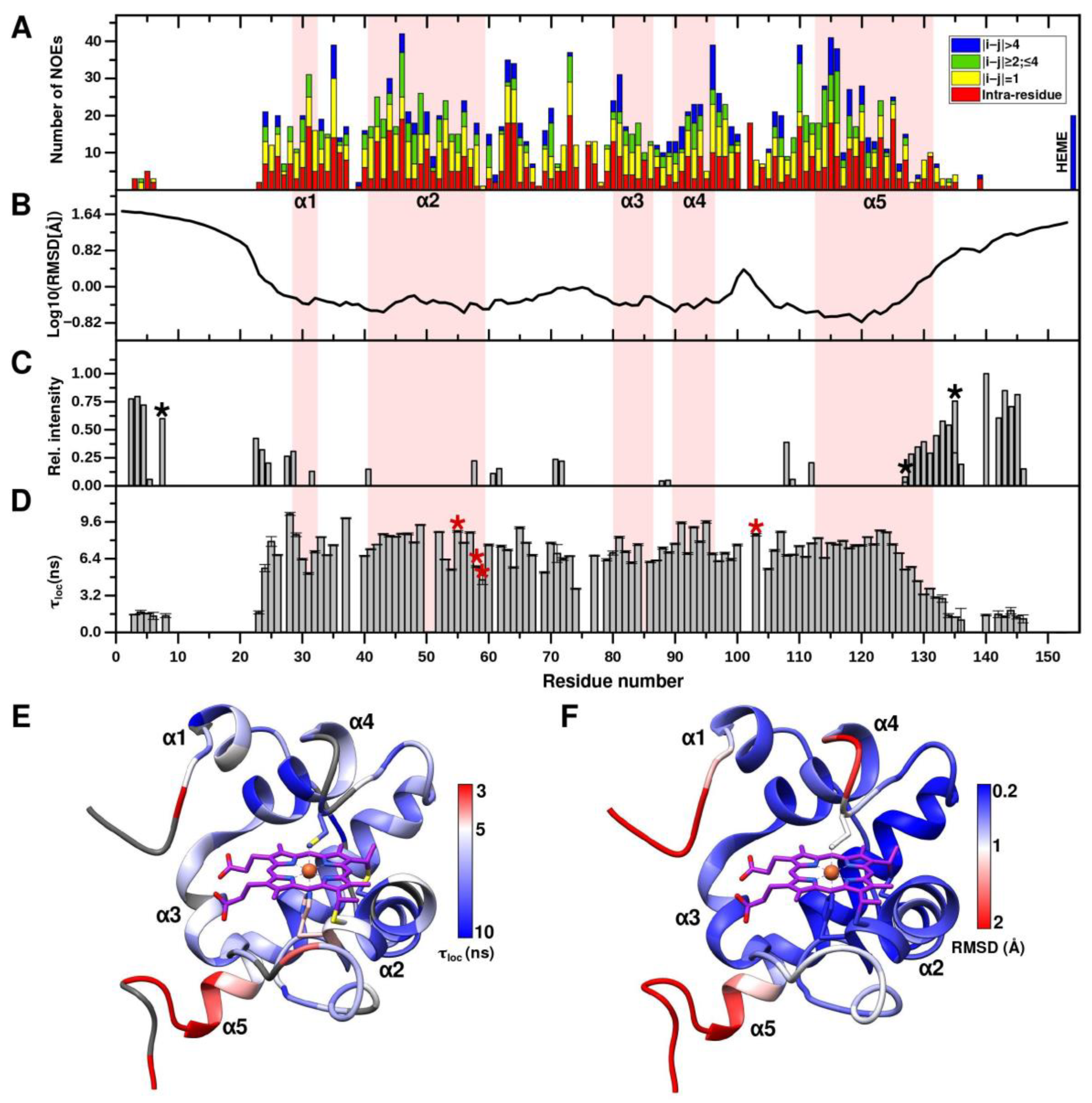
2.2. CytC552 Solution Structure Revealed Significant Heme Exposure to Solvent and Disordered N- and C-Terminal Extensions
2.3. Axial Heme Methionine Fluxionality of CytC552
2.4. NMR CSP Analysis of CytC552 Interaction with TcDH
2.5. CytC552-TcDH Complex Modeling and Electron Transfer Pathways Prediction
3. Materials and Methods
3.1. Purification of CytC552 from the Periplasmic Fraction of the T. paradoxus ARh1 Cells
3.2. Production of Recombinant CytC552
3.3. Activity Assays
3.4. Potentiometric Titration
3.5. Determination of the Dissociation Constant (Kd) of the TcDH-CytC552 Complex by ITC
3.6. NMR Spectroscopy
3.7. CytC552 NMR Solution Structure Calculation
3.8. SAXS Measurement and Analysis
3.9. HADDOCK Modeling of the TcDH-CytC552 Complex and Electron Transfer (ET) Pathways Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sorokin, D.Y.; Tourova, T.P.; Lysenko, A.M.; Kuenen, J.G. Microbial thiocyanate utilization under highly alkaline conditions. Appl. Environ. Microbiol. 2001, 67, 528–538. [Google Scholar] [CrossRef]
- Berben, T.; Overmars, L.; Sorokin, D.Y.; Muyzer, G. Comparative genome analysis of three thiocyanate oxidizing Thioalkalivibrio species isolated from soda lakes. Front. Microbiol. 2017, 8, 254. [Google Scholar] [CrossRef]
- Tikhonova, T.V.; Sorokin, D.Y.; Hagen, W.R.; Khrenova, M.G.; Muyzer, G.; Rakitina, T.V.; Shabalin, I.G.; Trofimov, A.A.; Tsallagov, S.I.; Popov, V.O. Trinuclear copper biocatalytic center forms an active site of thiocyanate dehydrogenase. Proc. Natl. Acad. Sci. USA 2020, 117, 5280–5290. [Google Scholar] [CrossRef]
- Stott, M.B.; Franzmann, P.D.; Zappia, L.R.; Watling, H.R.; Quan, L.P.; Clark, B.J.; Houchin, M.R.; Miller, P.C.; Williams, T.L. Thiocyanate removal from saline CIP process water by a rotating biological contactor, with reuse of the water for bioleaching. Hydrometallurgy 2001, 62, 93–105. [Google Scholar] [CrossRef]
- Dash, R.R.; Gaur, A.; Balomajumder, C. Cyanide in industrial wastewaters and its removal: A review on biotreatment. J. Hazard. Mater. 2009, 163, 1–11. [Google Scholar] [CrossRef]
- Gould, W.D.; King, M.; Mohapatra, B.R.; Cameron, R.A.; Kapoor, A.; Koren, D. A critical review on destruction of thiocyanate in mining effluents. Miner. Eng. 2012, 34, 38–47. [Google Scholar] [CrossRef]
- Kosohin, O.; Makohoniuk, O.; Kushmyruk, A. Electrochemical Oxidation of Thiocyanate on Metal Oxide Electrodes. Mater. Today Proc. 2019, 6, 219–226. [Google Scholar] [CrossRef]
- Collado, S.; Laca, A.; Díaz, M. Catalytic wet oxidation of thiocyanate with homogeneous copper(II) sulphate catalyst. J. Hazard. Mater. 2010, 177, 183–189. [Google Scholar] [CrossRef]
- Spurr, L.P.; Watts, M.P.; Gan, H.M.; Moreau, J.W. Biodegradation of thiocyanate by a native groundwater microbial consortium. PeerJ 2019, 7, e6498. [Google Scholar] [CrossRef]
- Prudencio, M.; Ubbink, M. Transient complexes of redox proteins: Structural and dynamic details from NMR studies. J. Mol. Recognit. 2004, 17, 524–539. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein—Protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Bonvin, A.M.J.J. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Balabin, I.A.; Hu, X.; Beratan, D.N. Exploring biological electron transfer pathway dynamics with the pathways plugin for VMD. J. Comput. Chem. 2012, 33, 906–910. [Google Scholar] [CrossRef]
- Tsallagov, S.I.; Sorokin, D.Y.; Tikhonova, T.V.; Popov, V.O.; Muyzer, G. Comparative genomics of Thiohalobacter thiocyanaticus HRh1T and Guyparkeria sp. SCN-R1, halophilic chemolithoautotrophic Sulfur-Oxidizing gammaproteobacteria capable of using thiocyanate as energy source. Front. Microbiol. 2019, 10, 898. [Google Scholar] [CrossRef]
- Bertini, I.; Cavallaro, G.; Rosato, A. Cytochrome c: Occurrence and functions. Chem. Rev. 2006, 106, 90–115. [Google Scholar] [CrossRef]
- Tiwari, P.; Kaila, P.; Guptasarma, P. Understanding anomalous mobility of proteins on SDS-PAGE with special reference to the highly acidic extracellular domains of human E- and N-cadherins. Electrophoresis 2019, 40, 1273–1281. [Google Scholar] [CrossRef]
- Sreenathan, B.R.; Taylor, C.P. The insensitivity of the 695 nm band of horse heart ferricytochrome c to protein conformation. Biochem. Biophys. Res. Commun. 1971, 42, 1122–1126. [Google Scholar] [CrossRef]
- Kaminsky, L.S.; Miller, V.J.; Davison, A.J. Thermodynamic studies of the opening of the heme crevice of ferricytochrome c. Biochemistry 1973, 12, 2215–2221. [Google Scholar] [CrossRef]
- Barr, I.; Guo, F. Pyridine hemochromagen assay for determining the concentration of heme in purified protein solutions. Bio-protocology 2015, 5, e1594. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Holm, L. Using Dali for protein structure comparison. In Structural Bioinformatics; Humana: New York, NY, USA, 2020; pp. 29–42. [Google Scholar] [CrossRef]
- Williams, P.A.; Coates, L.; Mohammed, F.; Gill, R.; Erskine, P.T.; Wood, J.S.P.; Cooper, B.; Anthony, C. The 1.6 Å X-ray structure of the unusual c-type cytochrome, cytochrome cL, from the methylotrophic bacterium Methylobacterium extorquens. J. Mol. Biol. 2006, 357, 151. [Google Scholar] [CrossRef]
- Nojiri, M.; Hira, D.; Yamaguchi, K.; Okajima, T.; Tanizawa, K.; Suzuki, S. Crystal structures of cytochrome CL and methanol dehydrogenase from Hyphomicrobium denitrificans: Structural and mechanistic insights into interactions between the two proteins. Biochemistry 2006, 45, 3481–3492. [Google Scholar] [CrossRef]
- Xia, Z.X.; Dai, W.W.; He, Y.N.; White, S.A.; Mathews, F.S.; Davidson, V.L. X-ray structure of methanol dehydrogenase from Paracoccus denitrificans and molecular modeling of its interactions with cytochrome c-551i. J. Biol. Inorg. Chem. 2003, 8, 843–854. [Google Scholar] [CrossRef]
- Ghosh, S.; Dhanasingh, I.; Ryu, J.; Kim, S.W.; Lee, S.H. Crystal structure of cytochrome CL from the aquatic methylotrophic bacterium Methylophaga aminisulfidivorans MP T. J. Microbiol. Biotechnol. 2020, 30, 1261–1271. [Google Scholar] [CrossRef]
- Afolabi, P.R.; Mohammed, F.; Amaratunga, K.; Majekodunmi, O.; Dales, L.; Gill, R.; Thompson, D.; Cooper, B.; Wood, P.; Goodwin, M.; et al. Site-directed mutagenesis and X-ray crystallography of the PQQ-containing quinoprotein methanol dehydrogenase and its electron acceptor, cytochrome CL. Biochemistry 2001, 40, 9799–9809. [Google Scholar] [CrossRef]
- Williams, P.A.; Coates, L.; Mohammed, F.; Gill, R.; Erskine, P.T.; Coker, A.; Wood, S.P.; Anthony, C.; Cooper, J.B. The atomic resolution of methanol dehydrogenase from Methylobacterium extorquens. Acta Cryst. 2005, 61, 75–79. [Google Scholar] [CrossRef]
- Chen, Z.W.; Matsushita, K.; Yamashita, T.; Fujii, T.A.; Toyama, H.; Adachi, O.; Bellamy, H.D.; Mathews, F.S. Structure at 1.9 Å resolution of a quinohemoprotein alcohol dehydrogenase from Pseudomonas putida HK5. Structure 2002, 10, 837–849. [Google Scholar] [CrossRef]
- Bertini, I.; Luchinat, C.; Parigi, G. Solution NMR of Paramagnetic Molecules: Applications Metallobiomolecules and Models; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Banci, L.; Bertini, I.; Huber, J.G.; Spyroulias, G.A.; Turano, P. Solution structure of reduced horse heart cytochrome c. J. Biol. Inorg. Chem. 1999, 4, 21–31. [Google Scholar] [CrossRef]
- Zhong, L.; Wen, X.; Rabinowitz, T.M.; Russell, B.S.; Karan, E.F.; Bren, K.L. Heme axial methionine fluxionality in Hydrogenobacter thermophilus cytochrome c552. Proc. Natl. Acad. Sci. USA 2004, 101, 8637–8642. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Bren, K.L. Heme Axial Methionine Fluxion in Pseudomonas aeruginosa Asn64Gln cytochrome c551. Inorg. Chem. 2005, 44, 8587–8593. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Morrison, J.F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. BBA-Enzymol. 1969, 185, 269–286. [Google Scholar] [CrossRef]
- Londer, Y.Y. Expression of recombinant cytochromes c in E. coli. Methods Mol. Biol. 2011, 705, 123–150. [Google Scholar] [CrossRef]
- Arslan, E.; Schulz, H.; Zufferey, R.; Kunzler, P.; Thony-Meyer, L. Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem. Biophys. Res. Commun. 1998, 251, 744–747. [Google Scholar] [CrossRef]
- Francis, R.T., Jr.; Becker, R.R. Specific indication of hemoproteins in polyacrylamide gels using a double-staining process. Anal. Biochem. 1964, 136, 509–514. [Google Scholar] [CrossRef]
- Dobbin, P.S.; Butt, J.N.; Powell, A.K.; Reid, G.A.; Richardson, D.J. Characterization of a flavocytochrome that is induced during the anaerobic respiration of Fe3+ by Shewanella frigidimarina NCIMB400. Biochem. J. 1999, 342, 439–448. [Google Scholar] [CrossRef]
- Favier, A.; Brutscher, B. Recovering lost magnetization: Polarization enhancement in biomolecular NMR. J. Biomol. NMR 2011, 49, 9–15. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A.D. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Chill, J.H.; Louis, J.M.; Baber, J.L.; Bax, A. Measurement of 15N relaxation in the detergent-solubilized tetrameric KcsA potassium channel. J. Biomol. NMR 2006, 36, 123–136. [Google Scholar] [CrossRef]
- Bardiaux, B.; Malliavin, T.; Nilges, M. ARIA for Solution and Solid-State NMR, Protein NMR Techniques; Humana Press: Totowa, NJ, USA, 2012; pp. 453–483. [Google Scholar] [CrossRef]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Warren, G.L. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Cryst. D 1998, 54, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Brunger, A.T. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2007, 2, 2728–2733. [Google Scholar] [CrossRef] [PubMed]
- Habeck, M.; Rieping, W.; Linge, J.P.; Nigles, M. NOE Assignment with ARIA 2.0, NMR Techniques; Humana Press: Totowa, NJ, USA, 2004; pp. 379–402. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A., III; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Giammona, D.A. An Examination of Conformational Flexibility in Porphyrins and Bulky-Ligand Binding in Myoglobin. Ph.D. Thesis, University of California, Davis, CA, USA, 1984. [Google Scholar]
- Autenrieth, F.; Tajkhorshid, E.; Baudry, J.; Luthey-Schulten, Z. Classical force field parameters for the heme prosthetic group of cytochrome c. J. Comput. Chem. 2004, 25, 1613–1622. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, G.M. Using Xplor–NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating protein structures determined by structural genomics consortia. Proteins Struct. Funct. Bioinform. 2007, 66, 778–795. [Google Scholar] [CrossRef]
- Gore, S.; García, E.S.; Hendrickx, P.M.; Gutmanas, A.; Westbrook, J.D.; Yang, H.; Feng, Z.; Baskaran, K.; Berrisford, J.M.; Hudson, B.P.; et al. Validation of structures in the Protein Data Bank. Structure 2017, 25, 1916–1927. [Google Scholar] [CrossRef] [Green Version]
- Manalastas-Canto, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Cryst. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Hopkins, J.B.; Gillilan, R.E.; Skou, S. BioXTAS RAW: Improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Cryst. 2017, 50, 1545–1553. [Google Scholar] [CrossRef]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013, 105, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software X 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Britikov, V.V.; Bocharov, E.V.; Britikova, E.V.; Dergousova, N.I.; Kulikova, O.G.; Solovieva, A.Y.; Shipkov, N.S.; Varfolomeeva, L.A.; Tikhonova, T.V.; Timofeev, V.I.; et al. Unusual Cytochrome c552 from Thioalkalivibrio paradoxus: Solution NMR Structure and Interaction with Thiocyanate Dehydrogenase. Int. J. Mol. Sci. 2022, 23, 9969. https://doi.org/10.3390/ijms23179969
Britikov VV, Bocharov EV, Britikova EV, Dergousova NI, Kulikova OG, Solovieva AY, Shipkov NS, Varfolomeeva LA, Tikhonova TV, Timofeev VI, et al. Unusual Cytochrome c552 from Thioalkalivibrio paradoxus: Solution NMR Structure and Interaction with Thiocyanate Dehydrogenase. International Journal of Molecular Sciences. 2022; 23(17):9969. https://doi.org/10.3390/ijms23179969
Chicago/Turabian StyleBritikov, Vladimir V., Eduard V. Bocharov, Elena V. Britikova, Natalia I. Dergousova, Olga G. Kulikova, Anastasia Y. Solovieva, Nikolai S. Shipkov, Larisa A. Varfolomeeva, Tamara V. Tikhonova, Vladimir I. Timofeev, and et al. 2022. "Unusual Cytochrome c552 from Thioalkalivibrio paradoxus: Solution NMR Structure and Interaction with Thiocyanate Dehydrogenase" International Journal of Molecular Sciences 23, no. 17: 9969. https://doi.org/10.3390/ijms23179969
APA StyleBritikov, V. V., Bocharov, E. V., Britikova, E. V., Dergousova, N. I., Kulikova, O. G., Solovieva, A. Y., Shipkov, N. S., Varfolomeeva, L. A., Tikhonova, T. V., Timofeev, V. I., Shtykova, E. V., Altukhov, D. A., Usanov, S. A., Arseniev, A. S., Rakitina, T. V., & Popov, V. O. (2022). Unusual Cytochrome c552 from Thioalkalivibrio paradoxus: Solution NMR Structure and Interaction with Thiocyanate Dehydrogenase. International Journal of Molecular Sciences, 23(17), 9969. https://doi.org/10.3390/ijms23179969