
Coagulation Factor XIIIa and Activated Protein C Activate Platelets via GPVI and PAR1

 , , , ,
, , , ,  ,
,
Abstract
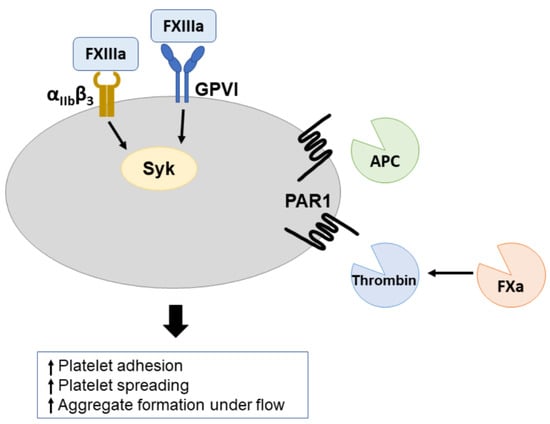
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Supernatant of Hirudin-Treated Coagulated Plasma Enhances Platelet Activation
2.2. Effect of Individual Coagulation Factors on Platelet Activation
2.3. Immobilization of APC and FXIIIa Favors Their Activating Effect on Platelets
2.3.1. Immobilized APC
2.3.2. Immobilized FXIIIa
2.4. Immobilized FXIIIa and APC Enhance Platelet Adhesion under Flow
3. Discussion
4. Materials and Methods
4.1. Preparation of Supernatant of Hirudin-Treated Coagulated Plasma (SCP)
4.2. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Heemskerk, J.W.; Mattheij, N.J.; Cosemans, J.M. Platelet-based coagulation: Different populations, different functions. J. Thromb. Haemost. 2013, 11, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Ollivier, V.; Loyau, S.; Schaff, M.; Dumont, B.; Favier, R.; Freyburger, G.; Latger-Cannard, V.; Nieswandt, B.; Gachet, C.; et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015, 126, 683–691. [Google Scholar] [CrossRef]
- Perrella, G.; Huang, J.; Provenzale, I.; Swieringa, F.; Heubel-Moenen, F.; Farndale, R.W.; Roest, M.; van der Meijden, P.E.J.; Thomas, M. Nonredundant Roles of Platelet Glycoprotein VI and Integrin alphaIIbbeta3 in Fibrin-Mediated Microthrombus Formation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e97–e111. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, O.M.; Hughes, C.E.; Montague, S.; Watson, S.K.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.U.; Kaneva, V.; Loyau, S.; Nechipurenko, D.; Receveur, N.; Le Bris, M.; Janus-Bell, E.; Didelot, M.; Rauch, A.; Susen, S.; et al. Pharmacological Blockade of Glycoprotein VI Promotes Thrombus Disaggregation in the Absence of Thrombin. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2127–2142. [Google Scholar] [CrossRef]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef]
- Jooss, N.J.; De Simone, I.; Provenzale, I.; Fernandez, D.I.; Brouns, S.L.N.; Farndale, R.W.; Henskens, Y.M.C.; Kuijpers, M.J.E.; Cate, H.T.; van der Meijden, P.E.J.; et al. Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces. Int. J. Mol. Sci. 2019, 20, 2788. [Google Scholar] [CrossRef]
- Petzold, T.; Thienel, M.; Dannenberg, L.; Mourikis, P.; Helten, C.; Ayhan, A.; M′Pembele, R.; Achilles, A.; Trojovky, K.; Konsek, D.; et al. Rivaroxaban Reduces Arterial Thrombosis by Inhibition of FXa-Driven Platelet Activation via Protease Activated Receptor-1. Circ. Res. 2020, 126, 486–500. [Google Scholar] [CrossRef]
- Al-Tamimi, M.; Grigoriadis, G.; Tran, H.; Paul, E.; Servadei, P.; Berndt, M.C.; Gardiner, E.E.; Andrews, R.K. Coagulation-induced shedding of platelet glycoprotein VI mediated by factor Xa. Blood 2011, 117, 3912–3920. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef]
- Magwenzi, S.G.; Ajjan, R.A.; Standeven, K.F.; Parapia, L.A.; Naseem, K.M. Factor XIII supports platelet activation and enhances thrombus formation by matrix proteins under flow conditions. J. Thromb. Haemost. 2011, 9, 820–833. [Google Scholar] [CrossRef]
- White, T.C.; Berny, M.A.; Tucker, E.I.; Urbanus, R.T.; de Groot, P.G.; Fernandez, J.A.; Griffin, J.H.; Gruber, A.; McCarty, O.J. Protein C supports platelet binding and activation under flow: Role of glycoprotein Ib and apolipoprotein E receptor 2. J. Thromb. Haemost. 2008, 6, 995–1002. [Google Scholar] [CrossRef]
- Baglia, F.A.; Gailani, D.; Lopez, J.A.; Walsh, P.N. Identification of a binding site for glycoprotein Ibalpha in the Apple 3 domain of factor XI. J. Biol. Chem. 2004, 279, 45470–45476. [Google Scholar] [CrossRef]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef]
- White-Adams, T.C.; Berny, M.A.; Tucker, E.I.; Gertz, J.M.; Gailani, D.; Urbanus, R.T.; de Groot, P.G.; Gruber, A.; McCarty, O.J. Identification of coagulation factor XI as a ligand for platelet apolipoprotein E receptor 2 (ApoER2). Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1602–1607. [Google Scholar] [CrossRef]
- Gibson, C.M.; Chakrabarti, A.K.; Mega, J.; Bode, C.; Bassand, J.P.; Verheugt, F.W.; Bhatt, D.L.; Goto, S.; Cohen, M.; Mohanavelu, S.; et al. Reduction of stent thrombosis in patients with acute coronary syndromes treated with rivaroxaban in ATLAS-ACS 2 TIMI 51. J. Am. Coll. Cardiol. 2013, 62, 286–290. [Google Scholar] [CrossRef]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without Aspirin in Stable Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef]
- Ruetzler, K.; Smilowitz, N.R.; Berger, J.S.; Devereaux, P.J.; Maron, B.A.; Newby, L.K.; Perez, V.d.J.; Sessler, D.I.; Wijeysundera, D.N. Diagnosis and Management of Patients With Myocardial Injury After Noncardiac Surgery: A Scientific Statement From the American Heart Association. Circulation 2021, 144, e287–e305. [Google Scholar] [CrossRef]
- Devereaux, P.J.; Duceppe, E.; Guyatt, G.; Tandon, V.; Rodseth, R.; Biccard, B.M.; Xavier, D.; Szczeklik, W.; Meyhoff, C.S.; Vincent, J.; et al. Dabigatran in patients with myocardial injury after non-cardiac surgery (MANAGE): An international, randomised, placebo-controlled trial. Lancet 2018, 391, 2325–2334. [Google Scholar] [CrossRef]
- De Ceunynck, K.; Peters, C.G.; Jain, A.; Higgins, S.J.; Aisiku, O.; Fitch-Tewfik, J.L.; Chaudhry, S.A.; Dockendorff, C.; Parikh, S.M.; Ingber, D.E.; et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc. Natl. Acad. Sci. USA 2018, 115, E982–E991. [Google Scholar] [CrossRef]
- Dardik, R.; Shenkman, B.; Tamarin, I.; Eskaraev, R.; Harsfalvi, J.; Varon, D.; Inbal, A. Factor XIII mediates adhesion of platelets to endothelial cells through alpha(v)beta(3) and glycoprotein IIb/IIIa integrins. Thromb. Res. 2002, 105, 317–323. [Google Scholar] [CrossRef]
- Cox, A.D.; Devine, D.V. Factor XIIIa binding to activated platelets is mediated through activation of glycoprotein IIb-IIIa. Blood 1994, 83, 1006–1016. [Google Scholar]
- Woodside, D.G.; Obergfell, A.; Leng, L.; Wilsbacher, J.L.; Miranti, C.K.; Brugge, J.S.; Shattil, S.J.; Ginsberg, M.H. Activation of Syk protein tyrosine kinase through interaction with integrin beta cytoplasmic domains. Curr. Biol. 2001, 11, 1799–1804. [Google Scholar] [CrossRef]
- Lee, T.Y.; Chang, C.C.; Lu, W.J.; Yen, T.L.; Lin, K.H.; Geraldine, P.; Li, J.Y.; Sheu, J.R. Honokiol as a specific collagen receptor glycoprotein VI antagonist on human platelets: Functional ex vivo and in vivo studies. Sci. Rep. 2017, 7, 40002. [Google Scholar] [CrossRef]
- Lecut, C.; Feeney, L.A.; Kingsbury, G.; Hopkins, J.; Lanza, F.; Gachet, C.; Villeval, J.L.; Jandrot-Perrus, M. Human platelet glycoprotein VI function is antagonized by monoclonal antibody-derived Fab fragments. J. Thromb. Haemost. 2003, 1, 2653–2662. [Google Scholar] [CrossRef]
- de Witt, S.M.; Swieringa, F.; Cavill, R.; Lamers, M.M.; van Kruchten, R.; Mastenbroek, T.; Baaten, C.; Coort, S.; Pugh, N.; Schulz, A.; et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat. Commun. 2014, 5, 4257. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Wilmen, A.; Wang, D.; Wang, X.; Bauzon, M.; Kim, J.Y.; Linden, L.; Li, L.; Egner, U.; Marquardt, T.; et al. Targeted inhibition of activated protein C by a non-active-site inhibitory antibody to treat hemophilia. Nat. Commun. 2020, 11, 2992. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wildhagen, K.C.; Schrijver, R.; Beckers, L.; Cate, H.t.; Reutelingsperger, C.P.; Lutgens, E.; Nicolaes, G.A. Effects of exogenous recombinant APC in mouse models of ischemia reperfusion injury and of atherosclerosis. PLoS ONE 2014, 9, e101446. [Google Scholar] [CrossRef] [Green Version]
- Soh, U.J.; Trejo, J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through beta-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. USA 2011, 108, E1372–E1380. [Google Scholar] [CrossRef] [PubMed]
- Aisiku, O.; Peters, C.G.; De Ceunynck, K.; Ghosh, C.C.; Dilks, J.R.; Fustolo-Gunnink, S.F.; Huang, M.; Dockendorff, C.; Parikh, S.M.; Flaumenhaft, R. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood 2015, 125, 1976–1985. [Google Scholar] [CrossRef] [PubMed]
- Moroi, M.; Induruwa, I.; Farndale, R.W.; Jung, S.M. Factor XIII is a newly identified binding partner for platelet collagen receptor GPVI-dimer-An interaction that may modulate fibrin crosslinking. Res. Pract. Thromb. Haemost. 2022, 6, e12697. [Google Scholar] [CrossRef]
- Poulter, N.S.; Pollitt, A.Y.; Owen, D.M.; Gardiner, E.E.; Andrews, R.K.; Shimizu, H.; Ishikawa, D.; Bihan, D.; Farndale, R.W.; Moroi, M.; et al. Clustering of glycoprotein VI (GPVI) dimers upon adhesion to collagen as a mechanism to regulate GPVI signaling in platelets. J. Thromb. Haemost. 2017, 15, 549–564. [Google Scholar] [CrossRef]
- Pallini, C.; Pike, J.A.; O’Shea, C.; Andrews, R.K.; Gardiner, E.E.; Watson, S.P.; Poulter, N.S. Immobilized collagen prevents shedding and induces sustained GPVI clustering and signaling in platelets. Platelets 2021, 32, 59–73. [Google Scholar] [CrossRef]
- Lorand, L. Factor XIII: Structure, activation, and interactions with fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001, 936, 291–311. [Google Scholar] [CrossRef]
- Mitchell, J.L.; Lionikiene, A.S.; Fraser, S.R.; Whyte, C.S.; Booth, N.A.; Mutch, N.J. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood 2014, 124, 3982–3990. [Google Scholar] [CrossRef]
- Zahid, M.; Mangin, P.; Loyau, S.; Hechler, B.; Billiald, P.; Gachet, C.; Jandrot-Perrus, M. The future of glycoprotein VI as an antithrombotic target. J. Thromb. Haemost. 2012, 10, 2418–2427. [Google Scholar] [CrossRef]
- Gilio, K.; Munnix, I.C.; Mangin, P.; Cosemans, J.M.; Feijge, M.A.; van der Meijden, P.E.; Olieslagers, S.; Chrzanowska-Wodnicka, M.B.; Lillian, R.; Schoenwaelder, S.; et al. Non-redundant roles of phosphoinositide 3-kinase isoforms alpha and beta in glycoprotein VI-induced platelet signaling and thrombus formation. J. Biol. Chem. 2009, 284, 33750–33762. [Google Scholar] [CrossRef]
- Veninga, A.; Baaten, C.; De Simone, I.; Tullemans, B.M.E.; Kuijpers, M.J.E.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Effects of Platelet Agonists and Priming on the Formation of Platelet Populations. Thromb. Haemost. 2021, 122, 726–738. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Simone, I.; Baaten, C.C.F.M.J.; Jandrot-Perrus, M.; Gibbins, J.M.; ten Cate, H.; Heemskerk, J.W.M.; Jones, C.I.; van der Meijden, P.E.J. Coagulation Factor XIIIa and Activated Protein C Activate Platelets via GPVI and PAR1. Int. J. Mol. Sci. 2022, 23, 10203. https://doi.org/10.3390/ijms231810203
De Simone I, Baaten CCFMJ, Jandrot-Perrus M, Gibbins JM, ten Cate H, Heemskerk JWM, Jones CI, van der Meijden PEJ. Coagulation Factor XIIIa and Activated Protein C Activate Platelets via GPVI and PAR1. International Journal of Molecular Sciences. 2022; 23(18):10203. https://doi.org/10.3390/ijms231810203
Chicago/Turabian StyleDe Simone, Ilaria, Constance C. F. M. J. Baaten, Martine Jandrot-Perrus, Jonathan M. Gibbins, Hugo ten Cate, Johan W. M. Heemskerk, Chris I. Jones, and Paola E. J. van der Meijden. 2022. "Coagulation Factor XIIIa and Activated Protein C Activate Platelets via GPVI and PAR1" International Journal of Molecular Sciences 23, no. 18: 10203. https://doi.org/10.3390/ijms231810203
APA StyleDe Simone, I., Baaten, C. C. F. M. J., Jandrot-Perrus, M., Gibbins, J. M., ten Cate, H., Heemskerk, J. W. M., Jones, C. I., & van der Meijden, P. E. J. (2022). Coagulation Factor XIIIa and Activated Protein C Activate Platelets via GPVI and PAR1. International Journal of Molecular Sciences, 23(18), 10203. https://doi.org/10.3390/ijms231810203