
Biochemical and Neuropathological Findings in a Creutzfeldt–Jakob Disease Patient with the Rare Val180Ile-129Val Haplotype in the Prion Protein Gene

 , ,
, ,  , , and
, , and 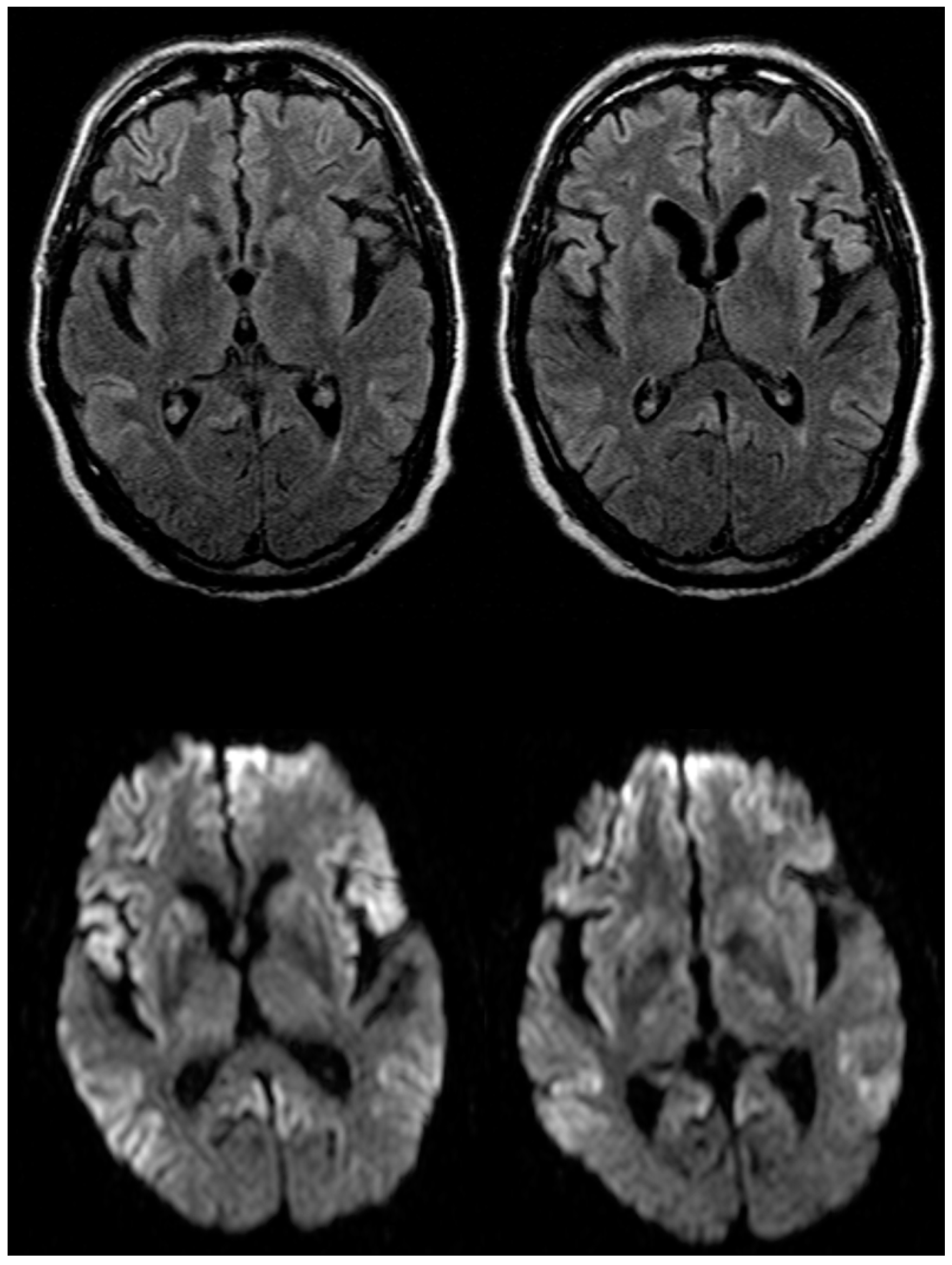{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Case Report
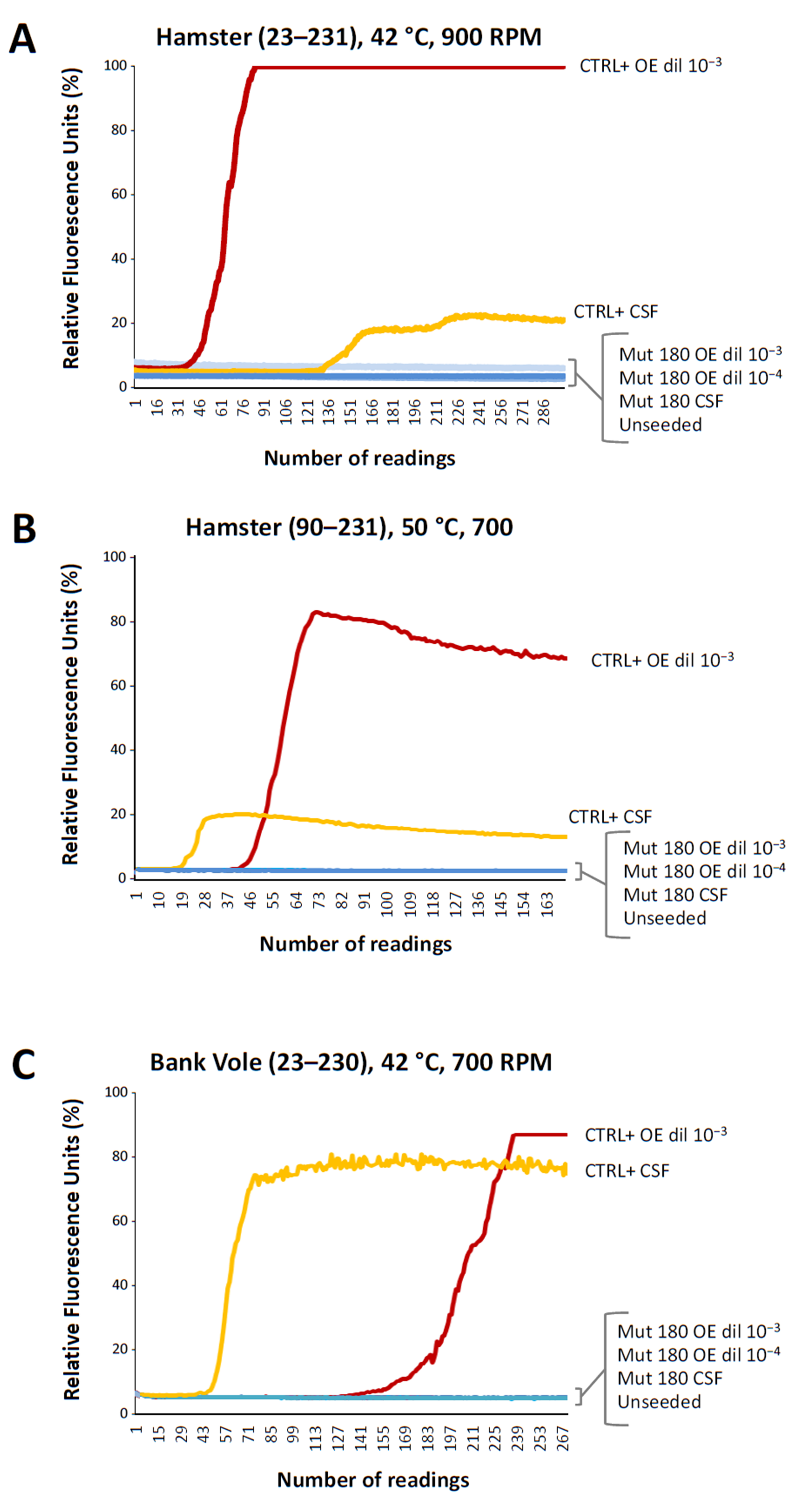
2.2. Real-Time Quaking Induced Conversion
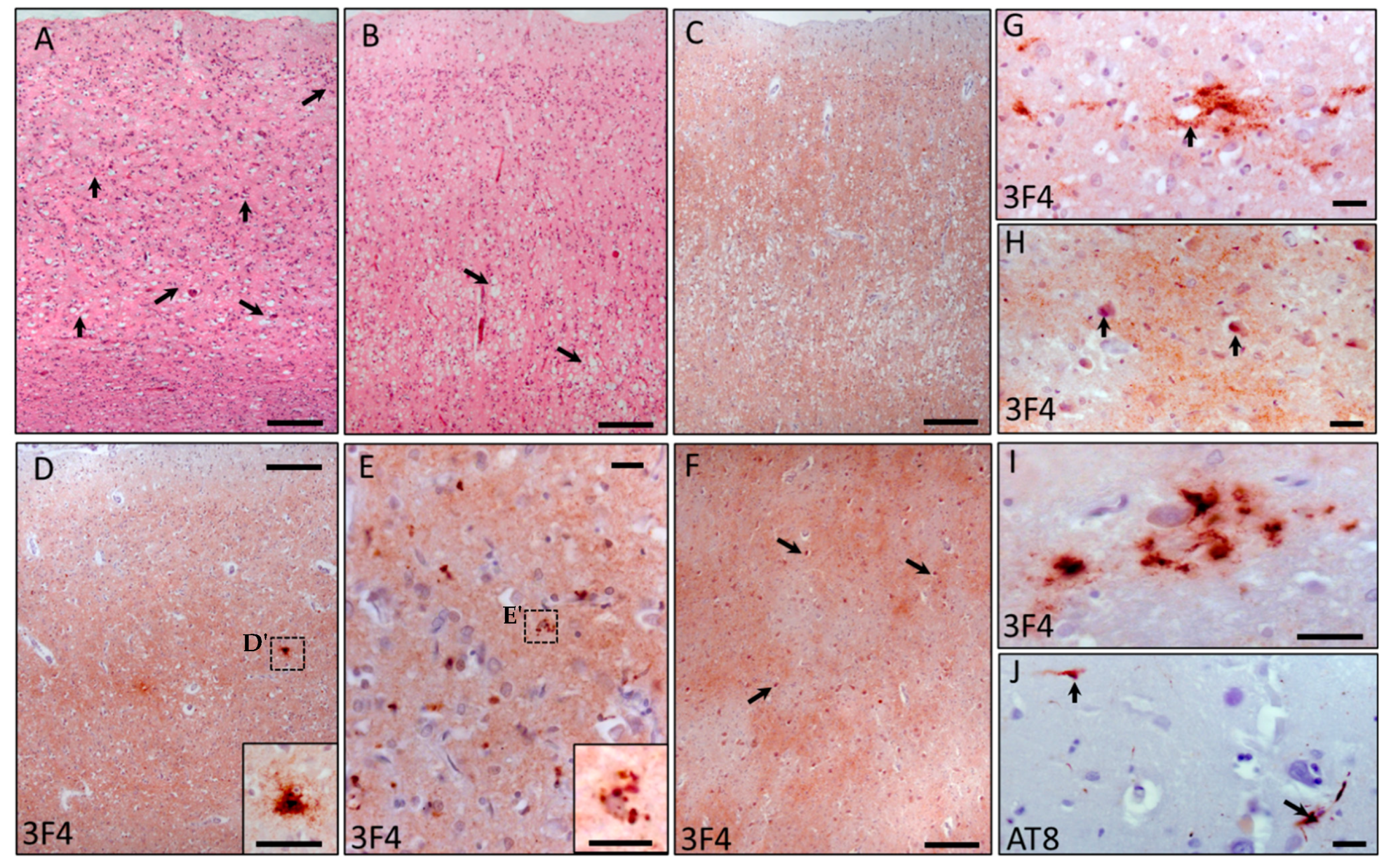
2.3. Neuropathological Changes
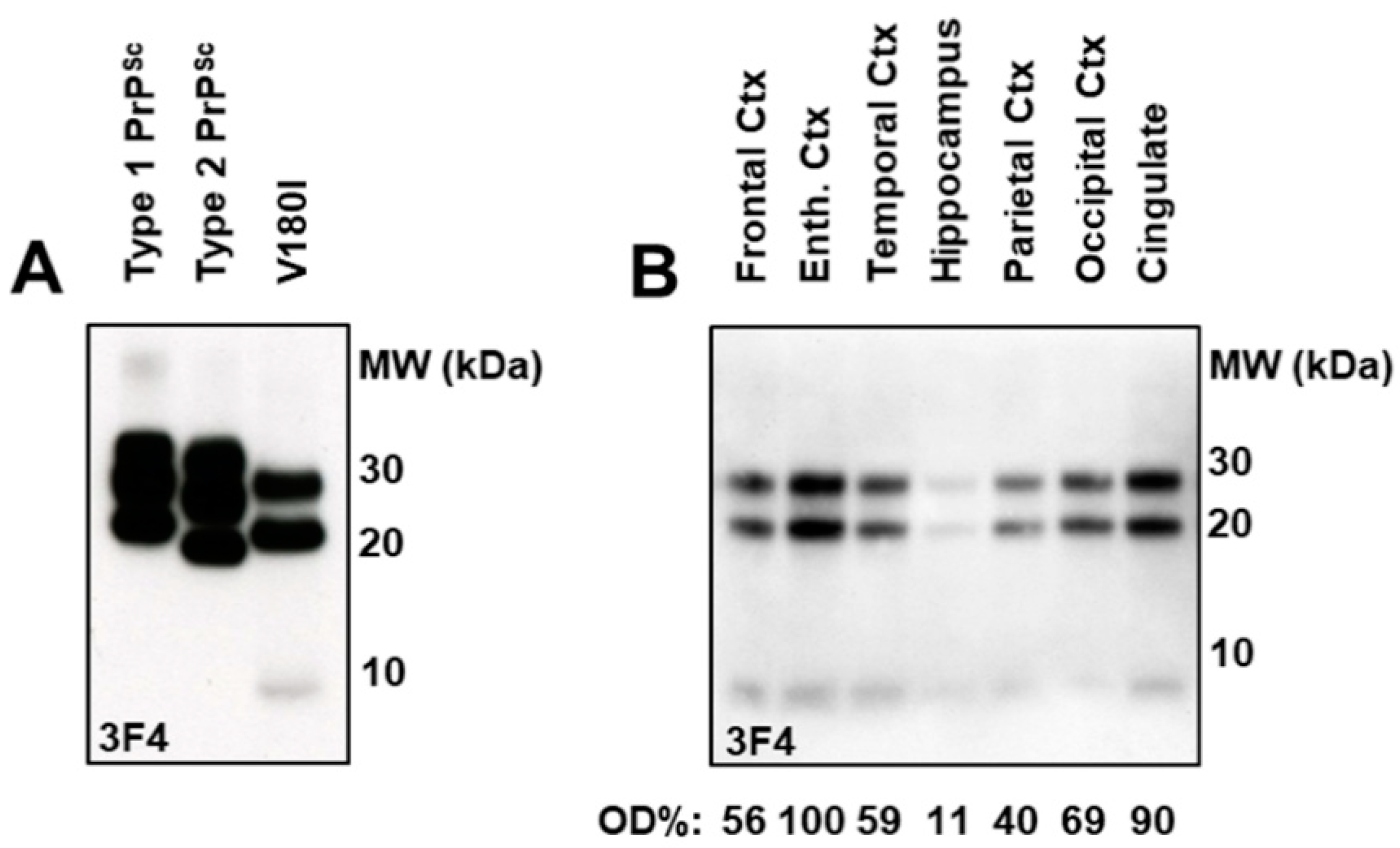
2.4. Immunoblot Analysis and Regional Distribution of Protease-Resistant PrP
3. Discussion
4. Materials and Methods
4.1. Real-Time Quaking Induced Conversion
4.2. Neuropathological Study
4.3. Western Blot Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ladogana, A.; Kovacs, G.G. Genetic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 219–242. [Google Scholar] [PubMed]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of Sporadic Creutzfeldt-Jakob Disease Based on Molecular and Phenotypic Analysis of 300 Subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Capellari, S.; Strammiello, R.; Saverioni, D.; Kretzschmar, H.; Parchi, P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: Insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 2011, 121, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Dittmar, K.; Llorens, F.; Gelpi, E.; Ferrer, I.; Schulz-Schaeffer, W.J.; Zerr, I. Hereditary human prion diseases: An update. Mol. Neurobiol. 2017, 54, 4138–4149. [Google Scholar] [CrossRef]
- McGuire, L.I.; Poleggi, A.; Poggiolini, I.; Suardi, S.; Grznarova, K.; Shi, S.; de Vil, B.; Sarros, S.; Satoh, K.; Cheng, K.; et al. Cerebrospinal fluid real-time quaking-induced conversion is a robust and reliable test for sporadic Creutzfeldt-Jakob disease: An international study. Ann. Neurol. 2016, 80, 160–165. [Google Scholar] [CrossRef]
- Orrú, C.D.; Bongianni, M.; Tonoli, G.; Ferrari, S.; Hughson, A.G.; Groveman, B.R.; Fiorini, M.; Pocchiari, M.; Monaco, S.; Caughey, B.; et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N. Engl. J. Med. 2014, 371, 519–529. [Google Scholar] [CrossRef]
- Bongianni, M.; Orrù, C.; Groveman, B.R.; Sacchetto, L.; Fiorini, M.; Tonoli, G.; Triva, G.; Capaldi, S.; Testi, S.; Ferrari, S.; et al. Diagnosis of human prion disease using real-time quaking-induced conversion testing of olfactory mucosa and cerebrospinal fluid samples. JAMA Neurol. 2017, 74, 155–162. [Google Scholar] [CrossRef]
- Orrú, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar]
- Zanusso, G.; Polo, A.; Farinazzo, A.; Nonno, R.; Cardone, F.; Di Bari, M.; Ferrari, S.; Principe, S.; Gelati, M.; Fasoli, E.; et al. Novel prion protein conformation and glycotype in Creutzfeldt-Jakob disease. Arch. Neurol. 2007, 64, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matucci, A.; Zanusso, G.; Gelati, M.; Farinazzo, A.; Fiorini, M.; Ferrari, S.; Andrighetto, G.; Cestari, T.; Caramelli, M.; Negro, A.; et al. Analysis of mammalian scrapie protein by novel monoclonal antibodies recognizing distinct prion protein glycoforms: An immunoblot and immunohistochemical study at the light and electron microscopic levels. Brain Res. Bull. 2005, 65, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Puopolo, M.; Ladogana, A.; Pocchiari, M.; Budka, H.; van Duijn, C.; Collins, S.J.; Boyd, A.; Giulivi, A.; Coulthart, M.; et al. Genetic prion disease: The EUROCJD experience. Hum. Genet. 2005, 118, 166–174. [Google Scholar] [CrossRef]
- Chasseigneaux, S.; Haïk, S.; Laffont-Proust, I.; De Marco, O.; Lenne, M.; Brandel, J.P.; Hauw, J.J.; Laplanche, J.L.; Peoc’h, K. V180I mutation of the prion protein gene associated with atypical PrPSc glycosylation. Neurosci. Lett. 2006, 408, 165–169. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.K.M.; Josviak, N.D.; Batistela, M.S.; Santos, P.S.F.; Landemberger, M.C.; Ramina, R. First case of V180I rare mutation in a Brazilian patient with Creutzfeldt-Jakob disease. Prion 2017, 11, 465–468. [Google Scholar] [CrossRef]
- Nozaki, I.; Hamaguchi, T.; Sanjo, N.; Noguchi-Shinohara, M.; Sakai, K.; Nakamura, Y.; Sato, T.; Kitamoto, T.; Mizusawa, H.; Moriwaka, F.; et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain 2010, 133, 3043–3057. [Google Scholar] [CrossRef]
- Lee, S.M.; Chung, M.; Hyeon, J.W.; Jeong, S.W.; Ju, Y.R.; Kim, H.; Lee, J.; Kim, S.Y.; An, S.S.; Cho, S.B.; et al. Genomic characteristics of genetic Creutzfeldt-Jakob disease patients with V180I mutation anda with other neurodegenerative disorders. PLoS ONE 2016, 11, e0157540. [Google Scholar]
- Jeong, B.H.; Kim, Y.S. Genetic Studies in Human Prion Diseases. J. Korean Med. Sci. 2014, 29, 623–632. [Google Scholar] [CrossRef]
- Qina, T.; Sanjo, N.; Hizume, M.; Higuma, M.; Tomita, M.; Atarashi, R.; Satoh, K.; Nozaki, I.; Hamaguchi, T.; Nakamura, Y.; et al. Clinical features of genetic Creutzfeldt-Jakob disease with V180I mutation in the prion protein gene. BMJ Open 2014, 4, e004968. [Google Scholar] [CrossRef]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.C.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef]
- Lee, J.; Chang, I.; Yu, W. Atomic insights into the effects of pathological mutants through the disruption of hydrophobic core in the prion protein. Sci. Rep. 2019, 9, 19144. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Shiga, Y.; Shibuya, S.; Chida, K.; Sato, Y.; Konno, H.; Doh-ura, K.; Kitamoto, T.; Itoyama, Y. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology 2004, 62, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Watson, N.; Brandel, J.P.; Green, A.; Hermann, P.; Ladogana, A.; Lindsay, T.; Mackenzie, J.; Pocchiari, M.; Smith, C.; Zerr, I.; et al. The importance of ongoing international surveillance for Creutzfeldt-Jakob disease. Nat. Rev. Neurol. 2021, 17, 362–379. [Google Scholar] [PubMed]
- Watson, N.; Hermann, P.; Ladogana, A.; Denouel, A.; Baiardi, S.; Colaizzo, E.; Giaccone, G.; Glatzel, M.; Green, A.J.E.; Haïk, S.; et al. Validation of revised international Creutzfeldt-Jakob disease surveillance network diagnostic criteria for sporadic Creutzfeldt-Jakob disease. JAMA Netw. Open 2022, 5, e2146319. [Google Scholar] [CrossRef] [PubMed]
- Akagi, A.; Iwasaki, Y.; Mimuro, M.; Kitamoto, T.; Yamada, M.; Yoshida, M. Pathological progression of genetic Creutzfeldt-Jakob disease with a PrP V180I mutation. Prion 2018, 12, 54–62. [Google Scholar] [CrossRef]
- Ito, Y.; Sanjo, N.; Hizume, M.; Kobayashi, A.; Ohgami, T.; Satoh, K.; Hamaguchi, T.; Yamada, M.; Kitamoto, T.; Mizusawa, H.; et al. Biochemical features of genetic Creutzfeldt-Jakob disease with valine-to-isoleucine substitution at codon 180 on the prion protein gene. Biochem. Biophys. Res. Commun. 2018, 496, 1055–1061. [Google Scholar] [CrossRef]
- Xiao, X.; Yuan, J.; Haïk, S.; Cali, I.; Zhan, Y.; Moudjou, M.; Li, B.; Laplanche, J.L.; Laude, H.; Langeveld, J.; et al. Glycoform-selective prion formation in sporadic and familial forms of prion disease. PLoS ONE 2013, 8, e58786. [Google Scholar]
- Wang, Z.; Yuan, J.; Shen, P.; Abskharon, R.; Lang, Y.; Dang, J.; Adornato, A.; Xu, L.; Chen, J.; Feng, J.; et al. In vitro seeding activity of glycoform-deficient prions from variably protease-sensitive prionopathy and familial CJD associated with PrPV180I mutation. Mol. Neurobiol. 2019, 56, 5456–5470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, X.; Ding, M.; Yuan, J.; Foutz, A.; Moudjou, M.; Kitamoto, T.; Langeveld, J.P.M.; Cui, L.; Zou, W.Q.; et al. Further characterization of glycoform-selective prions of variably protease-sensitive prionopathy. Pathogens 2021, 10, 513. [Google Scholar] [CrossRef]
- Higuma, M.; Sanjo, N.; Satoh, K.; Shiga, Y.; Sakai, K.; Nozaki, I.; Hamaguchi, T.; Nakamura, Y.; Kitamoto, T.; Shirabe, S.; et al. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PLoS ONE 2013, 8, e60003. [Google Scholar] [CrossRef]
- Melis, M.; Molari, A.; Floris, G.; Vascellari, S.; Balestrino, L.; Ladogana, A.; Poleggi, A.; Parchi, P.; Cossu, G.; Melis, M.; et al. Genetic Creutzfeldt-Jakob disease in Sardinia: A case series linked to the PRNP R208H mutation due to a single founder effect. Neurogenetics 2020, 21, 251–257. [Google Scholar] [PubMed]
- Tateishi, J.; Kitamoto, T.; Hoque, M.Z.; Furukawa, H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology 1996, 46, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Arshad, H.; Bourkas, M.E.C.; Watts, J.C. The utility of bank voles for studying prion disease. Prog. Mol. Biol. Transl. Sci. 2020, 175, 179–211. [Google Scholar] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanusso, G.; Colaizzo, E.; Poleggi, A.; Masullo, C.; Romeo, R.; Ferrari, S.; Bongianni, M.; Fiorini, M.; Tiple, D.; Vaianella, L.; et al. Biochemical and Neuropathological Findings in a Creutzfeldt–Jakob Disease Patient with the Rare Val180Ile-129Val Haplotype in the Prion Protein Gene. Int. J. Mol. Sci. 2022, 23, 10210. https://doi.org/10.3390/ijms231810210
Zanusso G, Colaizzo E, Poleggi A, Masullo C, Romeo R, Ferrari S, Bongianni M, Fiorini M, Tiple D, Vaianella L, et al. Biochemical and Neuropathological Findings in a Creutzfeldt–Jakob Disease Patient with the Rare Val180Ile-129Val Haplotype in the Prion Protein Gene. International Journal of Molecular Sciences. 2022; 23(18):10210. https://doi.org/10.3390/ijms231810210
Chicago/Turabian StyleZanusso, Gianluigi, Elisa Colaizzo, Anna Poleggi, Carlo Masullo, Raffaello Romeo, Sergio Ferrari, Matilde Bongianni, Michele Fiorini, Dorina Tiple, Luana Vaianella, and et al. 2022. "Biochemical and Neuropathological Findings in a Creutzfeldt–Jakob Disease Patient with the Rare Val180Ile-129Val Haplotype in the Prion Protein Gene" International Journal of Molecular Sciences 23, no. 18: 10210. https://doi.org/10.3390/ijms231810210
APA StyleZanusso, G., Colaizzo, E., Poleggi, A., Masullo, C., Romeo, R., Ferrari, S., Bongianni, M., Fiorini, M., Tiple, D., Vaianella, L., Sbriccoli, M., Porreca, F., Equestre, M., Pocchiari, M., Cardone, F., & Ladogana, A. (2022). Biochemical and Neuropathological Findings in a Creutzfeldt–Jakob Disease Patient with the Rare Val180Ile-129Val Haplotype in the Prion Protein Gene. International Journal of Molecular Sciences, 23(18), 10210. https://doi.org/10.3390/ijms231810210