
Recent Advances in PROTACs for Drug Targeted Protein Research



Abstract
:1. Introduction
2. Application of PROTAC in Anticancer
2.1. Breast Cancer
2.2. Hematological Tumors
2.2.1. Leukemia
2.2.2. Malignant Lymphoma
2.3. Colon Cancer
2.4. Prostatic Cancer
2.5. Pancreatic Cancer
3. Application of PROTAC in Immune Diseases
3.1. IRAK3
3.2. IRAK4
3.3. HDAc3
3.4. HDAc6
3.5. HPGDs
3.6. IDo1
3.7. Sirt2
3.8. PCAF/GCN5
3.9. RIPk2
3.10. ASK1
4. Application of PROTAC in Neurodegenerative Diseases
4.1. Alzheimer’s Disease
4.2. Huntington’s Disease
4.3. Parkinson’s Disease
5. Application of PROTAC in Cardiovascular Diseases
6. Application of PROTAC in Antiviral
7. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Gu, S.; Cui, D.; Chen, X.; Xiong, X.; Zhao, Y. PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. BioEssays 2018, 40, e1700247. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Aguilar, A.; Liu, Z.; Yang, C.Y.; Wang, S. Simple Structural Modifications Converting a Bona fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem. 2019, 62, 9471–9487. [Google Scholar] [CrossRef]
- Tan, L.; Gray, N.S. When Kinases Meet PROTACs. Chin. J. Chem. 2018, 36, 971–977. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- Sakamoto, K.M. Protacs for treatment of cancer. Pediatr. Res. 2010, 67, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Ottis, P.; Crews, C.M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12, 892–898. [Google Scholar] [CrossRef]
- Nguyen, C.; West, G.M.; Geoghegan, K.F. Emerging Methods in Chemoproteomics with Relevance to Drug Discovery. Methods Mol. Biol. 2017, 1513, 11–22. [Google Scholar]
- Raina, K.; Crews, C.M. Chemical inducers of targeted protein degradation. J. Biol. Chem. 2010, 285, 11057–11060. [Google Scholar]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. Publisher Correction: BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 846. [Google Scholar] [CrossRef]
- Drummond, M.L.; Williams, C.I. In Silico Modeling of PROTAC-Mediated Ternary Complexes: Validation and Application. J. Chem. Inf. Modeling 2019, 59, 1634–1644. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Roy, M.J.; Winkler, S.; Hughes, S.J.; Whitworth, C.; Galant, M.; Farnaby, W.; Rumpel, K.; Ciulli, A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14, 361–368. [Google Scholar] [CrossRef]
- Hughes, S.J.; Ciulli, A. Molecular recognition of ternary complexes: A new dimension in the structure-guided design of chemical degraders. Essays Biochem. 2017, 61, 505–516. [Google Scholar]
- Riching, K.M.; Mahan, S.; Corona, C.R.; McDougall, M.; Vasta, J.D.; Robers, M.B.; Urh, M.; Daniels, D.L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13, 2758–2770. [Google Scholar] [CrossRef]
- Toure, M.; Crews, C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. Engl. 2016, 55, 1966–1973. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sun, L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef]
- Jevtić, P.; Haakonsen, D.L.; Rapé, M. An E3 ligase guide to the galaxy of small-molecule-induced protein degradation. Cell Chem. Biol. 2021, 28, 1000–1013. [Google Scholar] [CrossRef]
- Kannt, A.; Đikić, I. Expanding the arsenal of E3 ubiquitin ligases for proximity-induced protein degradation. Cell Chem. Biol. 2021, 28, 1014–1031. [Google Scholar] [CrossRef]
- Bond, M.J.; Crews, C.M. Proteolysis targeting chimeras (PROTACs) come of age: Entering the third decade of targeted protein degradation. RSC Chem. Biol. 2021, 2, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef]
- Benowitz, A.B.; Jones, K.L.; Harling, J.D. The therapeutic potential of PROTACs. Expert Opin. Ther. Pat. 2021, 31, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Zeng, S.; Huang, W.; Zheng, X.; Liyan, C.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Raina, K.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; et al. An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2018, 36, 381. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79, P5–P04. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.; Park, S.B. Privileged structures: Efficient chemical “navigators” toward unexplored biologically relevant chemical spaces. J. Am. Chem. Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef] [PubMed]
- Xi, M.; Chen, Y.; Yang, H.; Xu, H.; Du, K.; Wu, C.; Xu, Y.; Deng, L.; Luo, X.; Yu, L.; et al. Small molecule PROTACs in targeted therapy: An emerging strategy to induce protein degradation. Eur. J. Med. Chem. 2019, 174, 159–180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef]
- Okuhira, K.; Demizu, Y.; Hattori, T.; Ohoka, N.; Shibata, N.; Nishimaki-Mogami, T.; Okuda, H.; Kurihara, M.; Naito, M. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 2013, 104, 1492–1498. [Google Scholar] [CrossRef]
- Ohoka, N.; Morita, Y.; Nagai, K.; Shimokawa, K.; Ujikawa, O.; Fujimori, I.; Ito, M.; Hayase, Y.; Okuhira, K.; Shibata, N.; et al. Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor α degradation. J. Biol. Chem. 2018, 293, 6776–6790. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Cromm, P.M.; Samarasinghe, K.T.G.; Hines, J.; Crews, C.M. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. [Google Scholar] [CrossRef]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Spradlin, J.N.; Hu, X.; Ward, C.C.; Brittain, S.M.; Jones, M.D.; Ou, L.; To, M.; Proudfoot, A.; Ornelas, E.; Woldegiorgis, M.; et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019, 15, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.Y.; Wang, M.; Han, X.; Wang, S. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2019, 62, 448–466. [Google Scholar] [CrossRef]
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e417. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e9. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar]
- Min, J.; Mayasundari, A.; Keramatnia, F.; Jonchere, B.; Yang, S.W.; Jarusiewicz, J.; Actis, M.; Das, S.; Young, B.; Slavish, J.; et al. Phenyl-Glutarimides: Alternative Cereblon Binders for the Design of PROTACs. Angew. Chem. Int. Ed. 2021, 60, 26663–26670. [Google Scholar] [CrossRef] [PubMed]
- Imaide, S.; Riching, K.M.; Makukhin, N.; Vetma, V.; Whitworth, C.; Hughes, S.J.; Trainor, N.; Mahan, S.D.; Murphy, N.; Cowan, A.D.; et al. Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity. Nat. Chem. Biol. 2021, 17, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Thummuri, D.; He, Y.; Liu, X.; Zhang, P.; Zhou, D.; Zheng, G. Utilizing PROTAC technology to address the on-target platelet toxicity associated with inhibition of BCL-X(L). Chem. Commun. 2019, 55, 14765–14768. [Google Scholar] [CrossRef]
- Zhang, X.; Thummuri, D.; Liu, X.; Hu, W.; Zhang, P.; Khan, S.; Yuan, Y.; Zhou, D.; Zheng, G. Discovery of PROTAC BCL-X(L) degraders as potent anticancer agents with low on-target platelet toxicity. Eur. J. Med. Chem. 2020, 192, 112186. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 2016, 55, 807–810. [Google Scholar] [CrossRef]
- Demizu, Y.; Shibata, N.; Hattori, T.; Ohoka, N.; Motoi, H.; Misawa, T.; Shoda, T.; Naito, M.; Kurihara, M. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg. Med. Chem. Lett. 2016, 26, 4865–4869. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Miyamoto, N.; Nagai, K.; Shimokawa, K.; Sameshima, T.; Ohoka, N.; Hattori, T.; Imaeda, Y.; Nara, H.; Cho, N.; et al. Development of protein degradation inducers of oncogenic BCR-ABL protein by conjugation of ABL kinase inhibitors and IAP ligands. Cancer Sci. 2017, 108, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Koch, R.; Budamagunta, V.; Lv, D.; Khan, S.; Zhang, X.; Wiegand, J.S.; Zheng, G.; Weinstock, D.M.; Zhou, D. DT2216, a BCL-XL Proteolysis Targeting Chimera (PROTAC), Is a Potent Anti T-Cell Lymphoma Agent That Does Not Induce Significant Thrombocytopenia. Blood 2019, 134, 303. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, N.; Song, Y.; Yang, Z.; Liu, W.; Zhu, J.; Rao, Y. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 2019, 33, 2105–2110. [Google Scholar] [CrossRef]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Figueroa, S.; Buhimschi, A.D.; Toure, M.; Hines, J.; Crews, C.M. Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2020, 30, 126877. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Dobrovolsky, D.; Wang, E.S.; Morrow, S.; Leahy, C.; Faust, T.; Nowak, R.P.; Donovan, K.A.; Yang, G.; Li, Z.; Fischer, E.S.; et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 2019, 133, 952–961. [Google Scholar] [CrossRef]
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. USA 2018, 115, e7285–e7292. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.D.; Correa, M.; Nagy, M.A.; Alexander, M.; Plantevin, V.; Grant, V.; Whitefield, B.; Huang, D.; Kercher, T.; Harris, R.; et al. Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem. 2020, 63, 6648–6676. [Google Scholar] [CrossRef]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Robb, C.M.; Contreras, J.I.; Kour, S.; Taylor, M.A.; Abid, M.; Sonawane, Y.A.; Zahid, M.; Murry, D.J.; Natarajan, A.; Rana, S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580. [Google Scholar] [CrossRef]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar]
- Chen, L.; Chen, Y.; Zhang, C.; Jiao, B.; Liang, S.; Tan, Q.; Chai, H.; Yu, W.; Qian, Y.; Yang, H.; et al. Discovery of First-In-Class Potent and Selective Tropomyosin Receptor Kinase Degraders. J. Med. Chem. 2020, 63, 14562–14575. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.H.; Lee, D.H.; Lee, C.O.; Du Ha, J.; Park, C.H.; Hwang, J.Y. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun. 2018, 505, 542–547. [Google Scholar]
- Powell, C.E.; Gao, Y.; Tan, L.; Donovan, K.A.; Nowak, R.P.; Loehr, A.; Bahcall, M.; Fischer, E.S.; Jänne, P.A.; George, R.E.; et al. Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2018, 61, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Huo, J.; Gu, X.; Wang, Y.; Wu, C.; Zhang, Q.; Wang, W.; Liu, Y.; Liu, Y.; Zhou, X.; et al. Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. J. Med. Chem. 2021, 64, 7839–7852. [Google Scholar]
- Xiao, Z.; Song, S.; Chen, D.; van Merkerk, R.; van der Wouden, P.E.; Cool, R.H.; Quax, W.J.; Poelarends, G.J.; Melgert, B.N.; Dekker, F.J. Proteolysis Targeting Chimera (PROTAC) for Macrophage Migration Inhibitory Factor (MIF) Has Anti-Proliferative Activity in Lung Cancer Cells. Angew. Chem. Int. Ed. Engl. 2021, 60, 17514–17521. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef] [PubMed]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.T. Understanding the roles of FAK in cancer: Inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C. Inhibition of p38: Has the fat lady sung? Arthritis Rheum. 2009, 60, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Manfroni, G.; Cecchetti, V.; Barreca, M.L. A Comprehensive Structural Overview of p38α Mitogen-Activated Protein Kinase in Complex with ATP-Site and Non-ATP-Site Binders. ChemMedChem 2018, 13, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Escós, A.; Risco, A.; Alsina-Beauchamp, D.; Cuenda, A. p38γ and p38δ Mitogen Activated Protein Kinases (MAPKs), New Stars in the MAPK Galaxy. Front. Cell Dev. Biol. 2016, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Alevy, Y.G.; Patel, A.C.; Romero, A.G.; Patel, D.A.; Tucker, J.; Roswit, W.T.; Miller, C.A.; Heier, R.F.; Byers, D.E.; Brett, T.J.; et al. IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J. Clin. Investig. 2012, 122, 4555–4568. [Google Scholar] [CrossRef] [PubMed]
- Yurtsever, Z.; Scheaffer, S.M.; Romero, A.G.; Holtzman, M.J.; Brett, T.J. The crystal structure of phosphorylated MAPK13 reveals common structural features and differences in p38 MAPK family activation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 790–799. [Google Scholar] [CrossRef]
- Yurtsever, Z.; Patel, D.A.; Kober, D.L.; Su, A.; Miller, C.A.; Romero, A.G.; Holtzman, M.J.; Brett, T.J. First comprehensive structural and biophysical analysis of MAPK13 inhibitors targeting DFG-in and DFG-out binding modes. Biochim. Biophys. Acta 2016, 1860, 2335–2344. [Google Scholar] [CrossRef]
- Zeng, H.; Belanger, D.B.; Curran, P.J.; Shipps, G.W., Jr.; Miao, H.; Bracken, J.B.; Siddiqui, M.A.; Malkowski, M.; Wang, Y. Discovery of novel imidazo[1,2-a]pyrazin-8-amines as Brk/PTK6 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5870–5875. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Shilatifard, A. Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 2016, 30, 2021–2041. [Google Scholar] [CrossRef] [Green Version]
- Deschler, B.; Lübbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, B.; Zechel, C.; Garnier, J.M.; Lutz, Y.; Tora, L.; Pierrat, P.; Heery, D.; Gronemeyer, H.; Chambon, P.; Losson, R. The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. Embo J. 1995, 14, 2020–2033. [Google Scholar] [CrossRef]
- Tsai, W.W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef]
- Cui, Z.; Cao, W.; Li, J.; Song, X.; Mao, L.; Chen, W. TRIM24 overexpression is common in locally advanced head and neck squamous cell carcinoma and correlates with aggressive malignant phenotypes. PLoS ONE 2013, 8, e63887. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef]
- Li, H.; Sun, L.; Tang, Z.; Fu, L.; Xu, Y.; Li, Z.; Luo, W.; Qiu, X.; Wang, E. Overexpression of TRIM24 correlates with tumor progression in non-small cell lung cancer. PLoS ONE 2012, 7, e37657. [Google Scholar] [CrossRef]
- Liu, X.; Huang, Y.; Yang, D.; Li, X.; Liang, J.; Lin, L.; Zhang, M.; Zhong, K.; Liang, B.; Li, J. Overexpression of TRIM24 is associated with the onset and progress of human hepatocellular carcinoma. PLoS ONE 2014, 9, e85462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, J.; Dong, M.; Yu, H.; Dai, X.; Li, K. Knockdown of tripartite motif containing 24 by lentivirus suppresses cell growth and induces apoptosis in human colorectal cancer cells. Oncol. Res. 2014, 22, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef]
- Kandarpa, M.; Peterson, L.F.; Potu, H.; Ramappan, M.; Liu, Y.; Polk, A.; Wang, S.; Talpaz, M. Improved Anti-Leukemic Pre-Clinical Efficacy of a Protac Based MDM2 Degrader in a Large AML Cohort. Blood 2019, 134, 2670. [Google Scholar] [CrossRef]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef]
- Grunwald, M.R.; Levis, M.J. FLT3 inhibitors for acute myeloid leukemia: A review of their efficacy and mechanisms of resistance. Int. J. Hematol. 2013, 97, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Cortes, J.; Roboz, G.J.; Rao, N.; Arowojolu, O.; Stine, A.; Shiotsu, Y.; Shudo, A.; Akinaga, S.; Small, D.; et al. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood 2009, 113, 3938–3946. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Yang, J.; Stark, G.R. Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer. 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schäfer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.I.; et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 2013, 24, 167–181. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell. 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Ghoshal, A.; Yugandhar, D.; Srivastava, A.K. BET inhibitors in cancer therapeutics: A patent review. Expert Opin. Ther. Pat. 2016, 26, 505–522. [Google Scholar] [CrossRef]
- Chonghaile, T.N.; Roderick, J.E.; Glenfield, C.; Ryan, J.; Sallan, S.E.; Silverman, L.B.; Loh, M.L.; Hunger, S.P.; Wood, B.; DeAngelo, D.J.; et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014, 4, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [Green Version]
- Will, B.; Zhou, L.; Vogler, T.O.; Ben-Neriah, S.; Schinke, C.; Tamari, R.; Yu, Y.; Bhagat, T.D.; Bhattacharyya, S.; Barreyro, L.; et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood 2012, 120, 2076–2086. [Google Scholar] [CrossRef]
- Zhang, Q.; Khan, S.; Zhang, X.; Kuruvilla, V.M.; Ghotbaldini, S.; Wells, J.; Baran, N.; Cai, T.; Han, L.; Ferrando, A.; et al. Targeting BCL-XL By Protac DT2216 Effectively Eliminates Leukemia Cells in T-ALL Pre-Clinical Models. Blood 2019, 134, 3870. [Google Scholar] [CrossRef]
- Hantschel, O.; Superti-Furga, G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Warsch, W.; Eckelhart, E.; Kaupe, I.; Grebien, F.; Wagner, K.U.; Superti-Furga, G.; Sexl, V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat. Chem. Biol. 2012, 8, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.A.; Forsythe, K.; Druker, B.J.; Hammer, D.; Boettiger, D.; Pear, W.S. BCR-ABL-induced adhesion defects are tyrosine kinase-independent. Blood 2002, 99, 4122–4130. [Google Scholar] [CrossRef]
- Ichim, C.V. Kinase-independent mechanisms of resistance of leukemia stem cells to tyrosine kinase inhibitors. Stem Cells Transl. Med. 2014, 3, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Müller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.; Christie, A.L.; Crombie, J.L.; Palmer, A.C.; Plana, D.; Shigemori, K.; Morrow, S.N.; Van Scoyk, A.; Wu, W.; Brem, E.A.; et al. Biomarker-driven strategy for MCL1 inhibition in T-cell lymphomas. Blood 2019, 133, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Hamp, I.; O’Neill, T.J.; Plettenburg, O.; Krappmann, D. A patent review of MALT1 inhibitors (2013-present). Expert Opin. Ther. Pat. 2021, 31, 1079–1096. [Google Scholar] [CrossRef]
- Fontan, L.; Goldstein, R.; Casalena, G.; Durant, M.; Teater, M.R.; Wilson, J.; Phillip, J.; Xia, M.; Shah, S.; Us, I.; et al. Identification of MALT1 feedback mechanisms enables rational design of potent antilymphoma regimens for ABC-DLBCL. Blood 2021, 137, 788–800. [Google Scholar] [CrossRef]
- Fontan, L.; Hatcher, J.; Scott, D.; Qiao, Q.; Us, I.; Du, G.; Durant, M.; Wilson, J.; Wu, H.; Gray, N.; et al. Chemically Induced Degradation of MALT1 to Treat B-Cell Lymphomas. Blood 2019, 134, 2073. [Google Scholar] [CrossRef]
- Zhang, H.; Qiu, L. Chapter 8—Bruton’s Tyrosine Kinase (BTK) Inhibitors as Sensitizing Agents for Cancer Chemotherapy. In Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy; Chen, Z.-S., Yang, D.-H., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 109–124. [Google Scholar]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef]
- Nijhof, I.S.; van de Donk, N.; Zweegman, S.; Lokhorst, H.M. Current and New Therapeutic Strategies for Relapsed and Refractory Multiple Myeloma: An Update. Drugs 2018, 78, 19–37. [Google Scholar] [CrossRef]
- Labianca, R.; Beretta, G.D.; Kildani, B.; Milesi, L.; Merlin, F.; Mosconi, S.; Pessi, M.A.; Prochilo, T.; Quadri, A.; Gatta, G.; et al. Colon cancer. Crit. Rev. Oncol. Hematol. 2010, 74, 106–133. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Rucker, P.V.; Wang, Z.; Fan, Y.; Albaugh, P.; Chopiuk, G.; Gessier, F.; Sun, F.; Adrian, F.; Liu, G.; et al. (R)-2-Phenylpyrrolidine Substituted Imidazopyridazines: A New Class of Potent and Selective Pan-TRK Inhibitors. ACS Med. Chem. Lett. 2015, 6, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Molecular Mechanisms and Targeted Therapies Including Immunotherapy for Non-Small Cell Lung Cancer. Curr. Cancer Drug Targets 2019, 19, 595–630. [Google Scholar] [CrossRef]
- Soumoy, L.; Kindt, N.; Ghanem, G.; Saussez, S.; Journe, F. Role of Macrophage Migration Inhibitory Factor (MIF) in Melanoma. Cancers 2019, 11, 529. [Google Scholar] [CrossRef]
- Cavalli, E.; Ciurleo, R.; Petralia, M.C.; Fagone, P.; Bella, R.; Mangano, K.; Nicoletti, F.; Bramanti, P.; Basile, M.S. Emerging Role of the Macrophage Migration Inhibitory Factor Family of Cytokines in Neuroblastoma. Pathogenic Effectors and Novel Therapeutic Targets? Molecules 2020, 25, 1194. [Google Scholar] [CrossRef]
- Coleman, A.M.; Rendon, B.E.; Zhao, M.; Qian, M.W.; Bucala, R.; Xin, D.; Mitchell, R.A. Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, D-dopachrome tautomerase. J. Immunol. 2008, 181, 2330–2337. [Google Scholar] [CrossRef]
- Charan, M.; Das, S.; Mishra, S.; Chatterjee, N.; Varikuti, S.; Kaul, K.; Misri, S.; Ahirwar, D.K.; Satoskar, A.R.; Ganju, R.K. Macrophage migration inhibitory factor inhibition as a novel therapeutic approach against triple-negative breast cancer. Cell Death Dis. 2020, 11, 774. [Google Scholar] [CrossRef]
- Zhang, M.; Yan, L.; Kim, J.A. Modulating mammary tumor growth, metastasis and immunosuppression by siRNA-induced MIF reduction in tumor microenvironment. Cancer Gene Ther. 2015, 22, 463–474. [Google Scholar] [CrossRef]
- Oliveira, C.S.; de Bock, C.E.; Molloy, T.J.; Sadeqzadeh, E.; Geng, X.Y.; Hersey, P.; Zhang, X.D.; Thorne, R.F. Macrophage migration inhibitory factor engages PI3K/Akt signalling and is a prognostic factor in metastatic melanoma. BMC Cancer 2014, 14, 630. [Google Scholar] [CrossRef] [PubMed]
- Balogh, K.N.; Templeton, D.J.; Cross, J.V. Macrophage Migration Inhibitory Factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses. PLoS ONE 2018, 13, e0197702. [Google Scholar] [CrossRef]
- Nguyen-Nielsen, M.; Borre, M. Diagnostic and Therapeutic Strategies for Prostate Cancer. Semin. Nucl. Med. 2016, 46, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Tingstedt, B.; Andersson, B.; Holmquist, F.; Sturesson, C.; Williamsson, C.; Sasor, A.; Borg, D.; Bauden, M.; Andersson, R. Pancreatic cancer: Yesterday, today and tomorrow. Future Oncol. 2016, 12, 1929–1946. [Google Scholar] [CrossRef]
- Muvaffak, A.; Pan, Q.; Yan, H.; Fernandez, R.; Lim, J.; Dolinski, B.; Nguyen, T.T.; Strack, P.; Wu, S.; Chung, R.; et al. Evaluating TBK1 as a therapeutic target in cancers with activated IRF3. Mol. Cancer Res. 2014, 12, 1055–1066. [Google Scholar] [CrossRef]
- Su, L.C.; Xu, W.D.; Huang, A.F. IRAK family in inflammatory autoimmune diseases. Autoimmun. Rev. 2020, 19, 102461. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef]
- Zhang, Y.; Diao, N.; Lee, C.K.; Chu, H.W.; Bai, L.; Li, L. Neutrophils Deficient in Innate Suppressor IRAK-M Enhances Anti-tumor Immune Responses. Mol. Ther. 2020, 28, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Kesselring, R.; Glaesner, J.; Hiergeist, A.; Naschberger, E.; Neumann, H.; Brunner, S.M.; Wege, A.K.; Seebauer, C.; Köhl, G.; Merkl, S.; et al. IRAK-M Expression in Tumor Cells Supports Colorectal Cancer Progression through Reduction of Antimicrobial Defense and Stabilization of STAT3. Cancer Cell 2016, 29, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Degorce, S.L.; Tavana, O.; Banks, E.; Crafter, C.; Gingipalli, L.; Kouvchinov, D.; Mao, Y.; Pachl, F.; Solanki, A.; Valge-Archer, V.; et al. Discovery of Proteolysis-Targeting Chimera Molecules that Selectively Degrade the IRAK3 Pseudokinase. J. Med. Chem. 2020, 63, 10460–10473. [Google Scholar] [CrossRef]
- Nunes, J.; McGonagle, G.A.; Eden, J.; Kiritharan, G.; Touzet, M.; Lewell, X.; Emery, J.; Eidam, H.; Harling, J.D.; Anderson, N.A. Targeting IRAK4 for Degradation with PROTACs. ACS Med. Chem. Lett. 2019, 10, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fu, L.; Shen, B.; Liu, Y.; Wang, W.; Cai, X.; Kong, L.; Yan, Y.; Meng, R.; Zhang, Z.; et al. Assessing IRAK4 Functions in ABC DLBCL by IRAK4 Kinase Inhibition and Protein Degradation. Cell Chem. Biol. 2020, 27, 1500–1509.e13. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, Y.; Bai, G.; Tong, L.; Zhang, T.; Zhou, J.; Zhang, H.; Xie, H.; Ding, J.; Duan, W. Design, Synthesis, and Biological Evaluation of IRAK4-Targeting PROTACs. ACS Med. Chem. Lett. 2021, 12, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; de Weerd, S.; Chen, D.; Zwinderman, M.R.H.; van der Wouden, P.E.; Dekker, F.J. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 208, 112800. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gu, Z.; Lin, S.; Chen, D.; Wang, J.; Zhao, Y.; Li, Y.; Liu, T.; Li, Y.; Wang, Y.; et al. Attenuation of NLRP3 Inflammasome Activation by Indirubin-Derived PROTAC Targeting HDAC6. ACS Chem. Biol. 2021, 16, 2746–2751. [Google Scholar] [CrossRef]
- Yokoo, H.; Shibata, N.; Naganuma, M.; Murakami, Y.; Fujii, K.; Ito, T.; Aritake, K.; Naito, M.; Demizu, Y. Development of a Hematopoietic Prostaglandin D Synthase-Degradation Inducer. ACS Med. Chem. Lett. 2021, 12, 236–241. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, W.; Wang, Y.; Yao, D.; Ye, T.; Yao, Y.; Chen, B.; Liu, G.; Yang, X.; Wang, W.; et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm. Sin. B. 2020, 10, 1943–1953. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Lehotzky, A.; Szunyogh, S.; Oláh, J.; Hammelmann, S.; Wössner, N.; Robaa, D.; Einsle, O.; Sippl, W.; Ovádi, J.; et al. HaloTag-Targeted Sirtuin-Rearranging Ligand (SirReal) for the Development of Proteolysis-Targeting Chimeras (PROTACs) against the Lysine Deacetylase Sirtuin 2 (Sirt2)*. Chembiochem 2020, 21, 3371–3376. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Fillmore, M.C.; Miah, A.H.; Chapman, T.D.; Maller, C.; Roberts, E.J.; Davis, L.C.; Lewis, D.E.; Galwey, N.W.; Waddington, K.E.; et al. Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem. Biol. 2018, 13, 2862–2867. [Google Scholar] [CrossRef]
- Mares, A.; Miah, A.H.; Smith, I.E.D.; Rackham, M.; Thawani, A.R.; Cryan, J.; Haile, P.A.; Votta, B.J.; Beal, A.M.; Capriotti, C.; et al. Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Commun. Biol. 2020, 3, 140. [Google Scholar] [CrossRef]
- Chaudhary, D.; Robinson, S.; Romero, D.L. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J. Med. Chem. 2015, 58, 96–110. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- McElroy, W.T. Interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitors: An updated patent review (2016–2018). Expert Opin. Ther. Pat. 2019, 29, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef]
- Magupalli, V.G.; Negro, R.; Tian, Y.; Hauenstein, A.V.; Di Caprio, G.; Skillern, W.; Deng, Q.; Orning, P.; Alam, H.B.; Maliga, Z.; et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 2020, 369, eaas8995. [Google Scholar] [CrossRef]
- Lewis, R.A.; Soter, N.A.; Diamond, P.T.; Austen, K.F.; Oates, J.A.; Roberts, L.J., 2nd. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J. Immunol. 1982, 129, 1627–1631. [Google Scholar] [PubMed]
- Aritake, K.; Kado, Y.; Inoue, T.; Miyano, M.; Urade, Y. Structural and functional characterization of HQL-79, an orally selective inhibitor of human hematopoietic prostaglandin D synthase. J. Biol. Chem. 2006, 281, 15277–15286. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef]
- Godin-Ethier, J.; Hanafi, L.A.; Piccirillo, C.A.; Lapointe, R. Indoleamine 2,3-dioxygenase expression in human cancers: Clinical and immunologic perspectives. Clin. Cancer Res. 2011, 17, 6985–6991. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Alam, H.B.; Liu, B.; Bronson, R.T.; Nikolian, V.C.; Wu, E.; Chong, W.; Li, Y. Selective Inhibition of SIRT2 Improves Outcomes in a Lethal Septic Model. Curr. Mol. Med. 2015, 15, 634–641. [Google Scholar] [CrossRef]
- Eskandarian, H.A.; Impens, F.; Nahori, M.A.; Soubigou, G.; Coppée, J.Y.; Cossart, P.; Hamon, M.A. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 2013, 341, 1238858. [Google Scholar] [CrossRef]
- Park, S.H.; Zhu, Y.; Ozden, O.; Kim, H.S.; Jiang, H.; Deng, C.X.; Gius, D.; Vassilopoulos, A. SIRT2 is a tumor suppressor that connects aging, acetylome, cell cycle signaling, and carcinogenesis. Transl. Cancer Res. 2012, 1, 15–21. [Google Scholar] [PubMed]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar] [CrossRef]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072–1077. [Google Scholar] [CrossRef]
- Humphreys, P.G.; Bamborough, P.; Chung, C.W.; Craggs, P.D.; Gordon, L.; Grandi, P.; Hayhow, T.G.; Hussain, J.; Jones, K.L.; Lindon, M.; et al. Discovery of a Potent, Cell Penetrant, and Selective p300/CBP-Associated Factor (PCAF)/General Control Nonderepressible 5 (GCN5) Bromodomain Chemical Probe. J. Med. Chem. 2017, 60, 695–709. [Google Scholar] [CrossRef]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Feldstein, A.E. NASH: Novel therapeutic strategies targeting ASK1 in NASH. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 329–330. [Google Scholar] [CrossRef]
- Schuster-Gaul, S.; Geisler, L.J.; McGeough, M.D.; Johnson, C.D.; Zagorska, A.; Li, L.; Wree, A.; Barry, V.; Mikaelian, I.; Jih, L.J.; et al. ASK1 inhibition reduces cell death and hepatic fibrosis in an Nlrp3 mutant liver injury model. JCI Insight 2020, 5, e123294. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Ma, F.Y.; Nikolic-Paterson, D.J. ASK1: A new therapeutic target for kidney disease. Am. J. Physiol. Renal Physiol. 2016, 311, F373–F381. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.H.; Etchevers, K.; Yi, S.; Breckenridge, D.; Hepner, M.; Patel, U.; Ling, J.; Mathias, A. Pharmacokinetics, Safety, and Tolerability of Selonsertib, an Apoptosis Signal-Regulating Kinase 1 (ASK1) Inhibitor, Following First-in-Human Single and Multiple Ascending Doses in Healthy Subjects. Clin. Pharmacokinet. 2020, 59, 1109–1117. [Google Scholar] [CrossRef]
- Hof, P.R.; Morrison, J.H. The aging brain: Morphomolecular senescence of cortical circuits. Trends Neurosci. 2004, 27, 607–613. [Google Scholar] [CrossRef]
- Chu, T.T.; Gao, N.; Li, Q.Q.; Chen, P.G.; Yang, X.F.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef]
- Kargbo, R.B. Treatment of Alzheimer’s by PROTAC-Tau Protein Degradation. ACS Med. Chem. Lett. 2019, 10, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8, e45457. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, Q.; Jiang, T.; Li, S.; Ye, J.; Zheng, J.; Wang, X.; Liu, Y.; Deng, M.; Ke, D.; et al. A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics 2021, 11, 5279–5295. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, J.; Wang, Y.; Liu, X.; Xu, K.; Xu, J.; Feng, F.; Sun, H. PROTACs suppression of GSK-3β, a crucial kinase in neurodegenerative diseases. Eur. J. Med. Chem. 2021, 210, 112949. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules that Induce the Degradation of Huntingtin. Angew. Chem. Int. Ed. Engl. 2017, 56, 11530–11533. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Degradation of huntingtin mediated by a hybrid molecule composed of IAP antagonist linked to phenyldiazenyl benzothiazole derivative. Bioorg. Med. Chem. Lett. 2018, 28, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. PROTAC Compounds Targeting α-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med. Chem. Lett. 2020, 11, 1086–1087. [Google Scholar] [CrossRef] [PubMed]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Drouin-Ouellet, J.; Tirolo, C.; Pulvirenti, A.; Giugno, R.; Testa, N.; Caniglia, S.; Serapide, M.F.; Cisbani, G.; Barker, R.A.; et al. GSK-3β-induced Tau pathology drives hippocampal neuronal cell death in Huntington’s disease: Involvement of astrocyte-neuron interactions. Cell Death Dis. 2016, 7, e2206. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Sirerol-Piquer, M.; Gomez-Ramos, P.; Hernández, F.; Perez, M.; Morán, M.A.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J.; García-Verdugo, J.M. GSK3β overexpression induces neuronal death and a depletion of the neurogenic niches in the dentate gyrus. Hippocampus 2011, 21, 910–922. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar] [PubMed] [Green Version]
- Ozansoy, M.; Başak, A.N. The central theme of Parkinson’s disease: α-synuclein. Mol. Neurobiol. 2013, 47, 460–465. [Google Scholar] [CrossRef]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Yang, Y.; Zhao, Q.; Wu, Y.; Song, L.; Yang, H.; He, M.; Gao, H.; Song, B.L.; Luo, J.; et al. Degradation versus Inhibition: Development of Proteolysis-Targeting Chimeras for Overcoming Statin-Induced Compensatory Upregulation of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase. J. Med. Chem. 2020, 63, 4908–4928. [Google Scholar] [CrossRef]
- Luo, G.; Li, Z.; Lin, X.; Li, X.; Chen, Y.; Xi, K.; Xiao, M.; Wei, H.; Zhu, L.; Xiang, H. Discovery of an orally active VHL-recruiting PROTAC that achieves robust HMGCR degradation and potent hypolipidemic activity in vivo. Acta Pharm. Sin. B 2021, 11, 1300–1314. [Google Scholar] [CrossRef]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Schulze zur Wiesch, J.; Schmitz, H.; Borowski, E.; Borowski, P. The proteins of the Hepatitis C virus: Their features and interactions with intracellular protein phosphorylation. Arch. Virol. 2003, 148, 1247–1267. [Google Scholar] [CrossRef]
- De Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and COVID-19 Therapeutics. ChemMedChem 2020, 15, 907–932. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease for the Potential Treatment of COVID-19. bioRxiv 2021. [Google Scholar] [CrossRef]



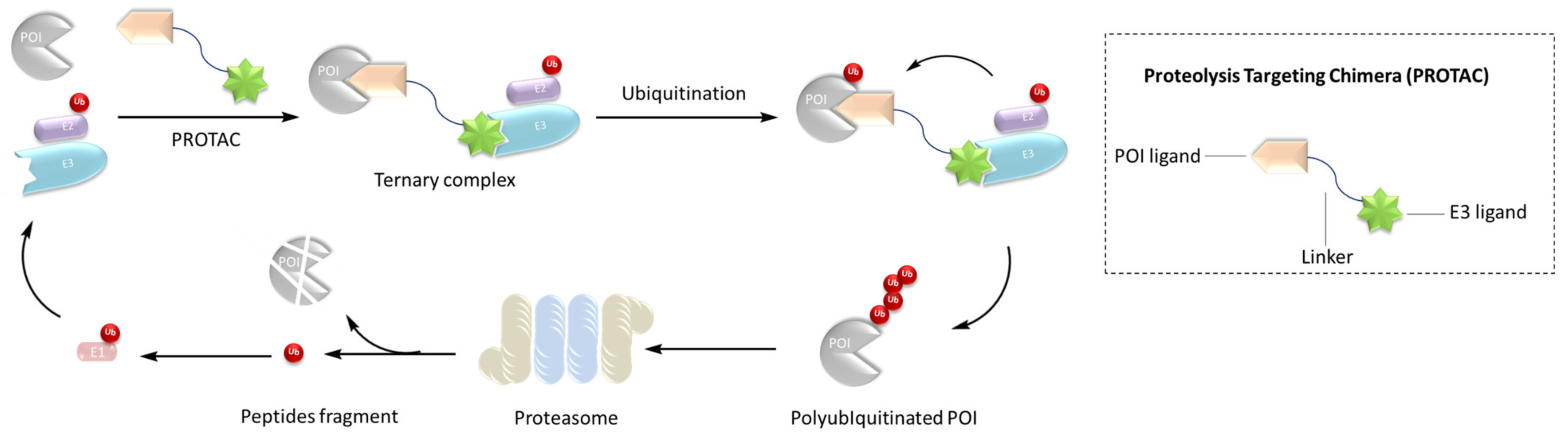{kind=link}
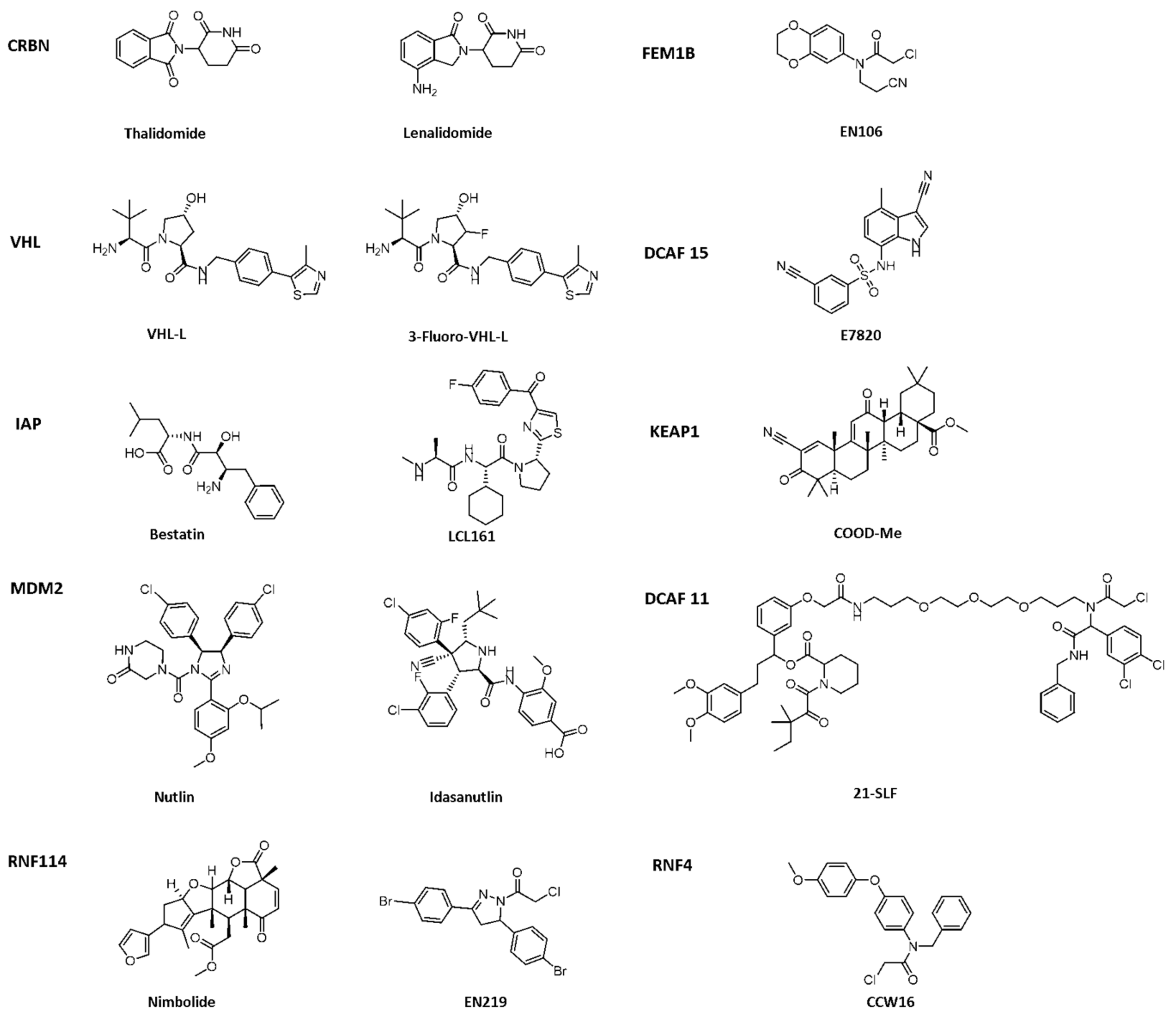{kind=link}
| Indication | PROTAC | Target | Structure | Activity | Ref. | |
|---|---|---|---|---|---|---|
| DC50 | Dmax% | |||||
| Breast cancer | 1 | ERα |  | - | - | [34] |
| 2 | ERRα |  | 100 nM | 86 | [36] | |
| 3 | FAK |  | 3 nM | 99 | [37] | |
| 4 | FAK |  | - | >90 | [38] | |
| 5 | p38α |  | 7.16 nM | 97.4 | [39] | |
| 6 | p38δ |  | 46 nM | 99.4 | [39] | |
| 7 | BCL-XL |  | 63 nM | 90.8 | [40] | |
| 8 | BRD4 |  | - | - | [41] | |
| 9 | PTK6 |  | - | - | [26] | |
| AML | 10 | RIPK2 |  | 1.4 nM | >95 | [36] |
| 11 | TRIM24 |  | - | - | [42] | |
| 12 | MDM2 |  | 1.5 nM | - | [43] | |
| 13 | FLT-3 |  | - | - | [44] | |
| 14 | STAT3 |  | - | >90 | [45] | |
| 15 | CDK6 |  | - | - | [46] | |
| 16 | BRD4 |  | 430 nM | - | [47] | |
| 17 | BRD4 |  | - | - | [15] | |
| 18 | BRD4 |  | <1 nM | 99 | [48] | |
| 19 | BRD4 |  | - | - | [49] | |
| T-ALL | 20 | BCL-XL |  | 50 nM | >85 | [50] |
| 21 | BCL-XL |  | 2.5 nM | - | [51] | |
| CML | 22 | BCR-ABL |  | - | >80 | [52] |
| 23 | BCR-ABL |  | - | - | [53] | |
| 24 | BCR-ABL |  | - | - | [54] | |
| TCL | 8 | BCL-XL |  | - | - | [55] |
| BCL | 25 | BTK |  | 29 nM | - | [56] |
| 26 | BTK |  | 6.2 nM | 99 | [57] | |
| 27 | BTK |  | 7.9 nM | 95 | [58] | |
| 28 | BTK |  | <1 nM | - | [59] | |
| 29 | BTK |  | - | - | [60] | |
| 30 | BTK |  | 5.9 nM | - | [61] | |
| MM | 31 | IKZF1-3 |  | - | - | [62] |
| 32 | CDK6 |  | 8.6 nM | - | [63] | |
| Colon cancer | 33 | ERK1-2 |  | - | - | [64] |
| 34 | CDK9 |  | - | - | [65] | |
| 35 | BRD4 |  | 32 nM | 98 | [66] | |
| 36 | TRK |  | 0.48 nM | - | [67] | |
| 37 | TRK |  | 0.36 nM | - | [67] | |
| NSCLC | 38 | ALK |  | - | - | [68] |
| 39 | ALK |  | 50 nM | - | [69] | |
| 40 | ALK |  | 50 nM | - | [69] | |
| 41 | EGFR; PARP |  | 0.47 μM | - | [70] | |
| 42 | MIF |  | 100 nM | >90 | [71] | |
| Prostatic cancer | 43 | AR |  | - | - | [72] |
| Pancreatic cancer | 44 | TBK1 |  | 32 nM | 96 | [73] |
| Protac | Target | Structure | Activity | Ref. | |
|---|---|---|---|---|---|
| DC50 | Dmax% | ||||
| 45 | IRAK3 |  | 2 nM | 98 | [149] |
| 46 | IRAK4 |  | 151 nM | - | [150] |
| 47 | IRAK4 |  | 405 nM | 90 | [151] |
| 48 | IRAK4 |  | - | - | [152] |
| 49 | HDAC3 |  | 0.32μM | - | [153] |
| 50 | HDAC3 |  | 42 nM | - | [154] |
| 51 | HDAC6 |  | 108.9 nM | 88 | [155] |
| 52 | H-PGDs |  | - | - | [156] |
| 53 | IDO1 |  | 2.84 μM | 93 | [157] |
| 54 | Sirt2 |  | - | - | [158] |
| 55 | Sirt2 |  | - | - | [159] |
| 56 | PCAF-GCN5 |  | 1.5–3 nM | >90 | [160] |
| 57 | RIPK2 |  | 1.4 nM | >95 | [36] |
| 58 | RIPK2 |  | 4 nM | - | [161] |
| 59 | RIPK2 |  | 2.5 nM | - | [161] |
| Indication | PROTAC | Target | Structure | Activity | Ref. | |
|---|---|---|---|---|---|---|
| DC50 | Dmax% | |||||
| AD | 60 | Tau |  | - | 75 | [188] |
| 61 | Tau |  | - | - | [189] | |
| 62 | GSK-3β |  | - | - | [190] | |
| HD | 63 | mHtt |  | - | - | [191] |
| 64 | mHtt |  | - | - | [191] | |
| 65 | mHtt |  | - | - | [192] | |
| PD | 66 | α-synuclein |  | - | 65 | [193] |
| Protac | Target | Structure | Activity | Ref. | |
|---|---|---|---|---|---|
| DC50 | Dmax% | ||||
| 67 | HMGCR |  | 0.1 μM | - | [202] |
| 68 |  | 120 nM | 76 | [203] | |
| 69 |  | - | 56 | [203] | |
| Indication | PROTAC | Target | Structure | Activity | Ref. | |
|---|---|---|---|---|---|---|
| DC50 | Dmax% | |||||
| HCV | 70 | NS3 |  | 50 nM | - | [206] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, T.; Xiao, H.; Wang, H.; Xu, X. Recent Advances in PROTACs for Drug Targeted Protein Research. Int. J. Mol. Sci. 2022, 23, 10328. https://doi.org/10.3390/ijms231810328
Yao T, Xiao H, Wang H, Xu X. Recent Advances in PROTACs for Drug Targeted Protein Research. International Journal of Molecular Sciences. 2022; 23(18):10328. https://doi.org/10.3390/ijms231810328
Chicago/Turabian StyleYao, Tingting, Heng Xiao, Hong Wang, and Xiaowei Xu. 2022. "Recent Advances in PROTACs for Drug Targeted Protein Research" International Journal of Molecular Sciences 23, no. 18: 10328. https://doi.org/10.3390/ijms231810328
APA StyleYao, T., Xiao, H., Wang, H., & Xu, X. (2022). Recent Advances in PROTACs for Drug Targeted Protein Research. International Journal of Molecular Sciences, 23(18), 10328. https://doi.org/10.3390/ijms231810328