
SARS-CoV-2 Spike Proteins and Cell–Cell Communication Inhibits TFPI and Induces Thrombogenic Factors in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for COVID-19 Coagulopathy Pathogenesis
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
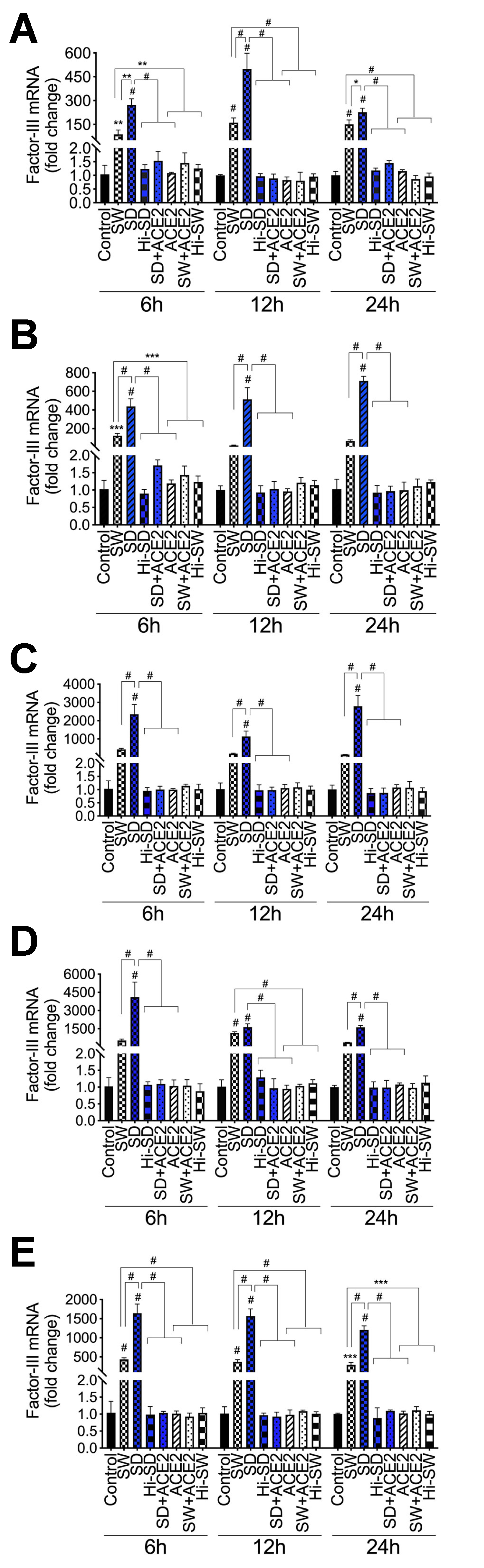
2.1. Exposure of HLMEC and Neutrophils to S-Proteins and Endothelial–Neutrophil Interactions Induced TF (Factor-III) Transcriptional Upregulation
2.2. Delta-Variant S-Proteins Induced Higher TF Levels Compared to Wuhan-Variant S-Proteins
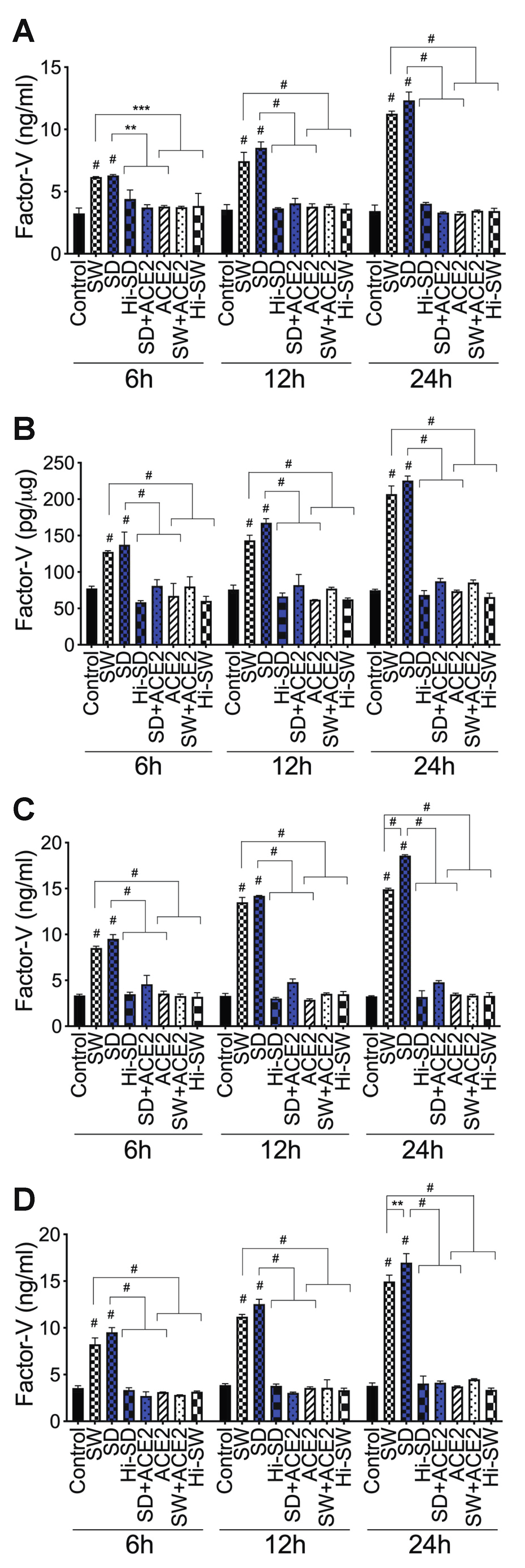
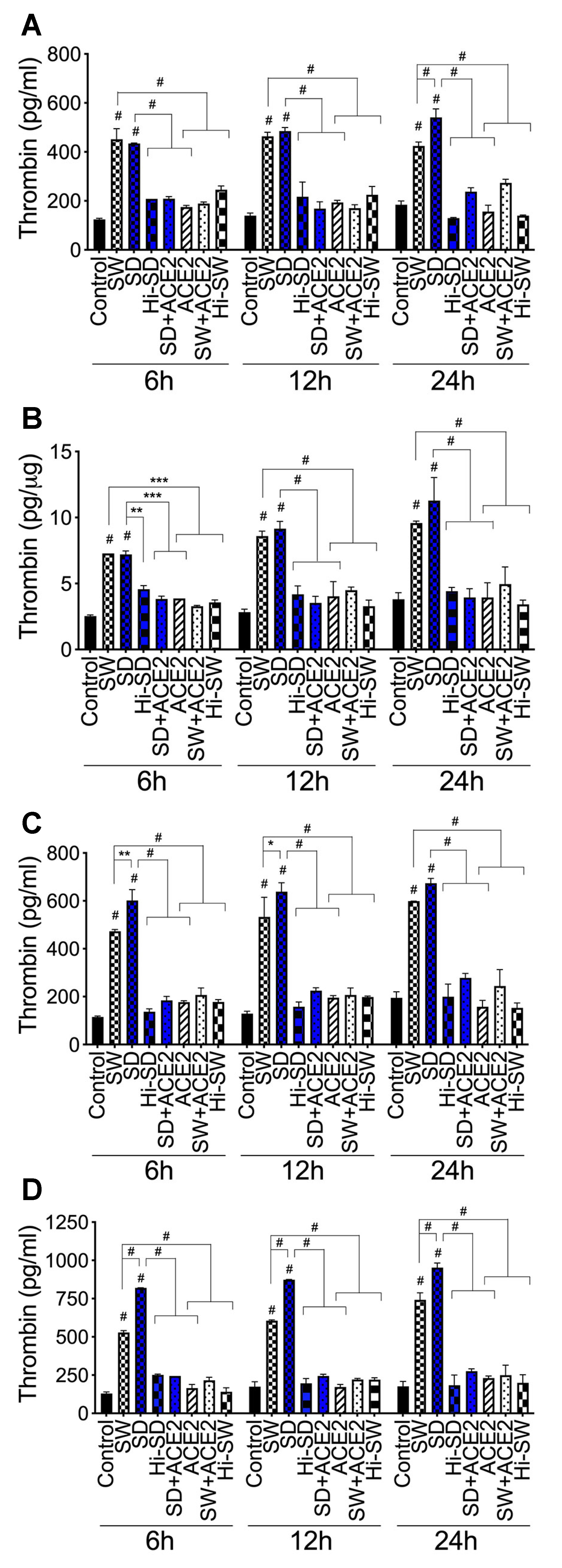
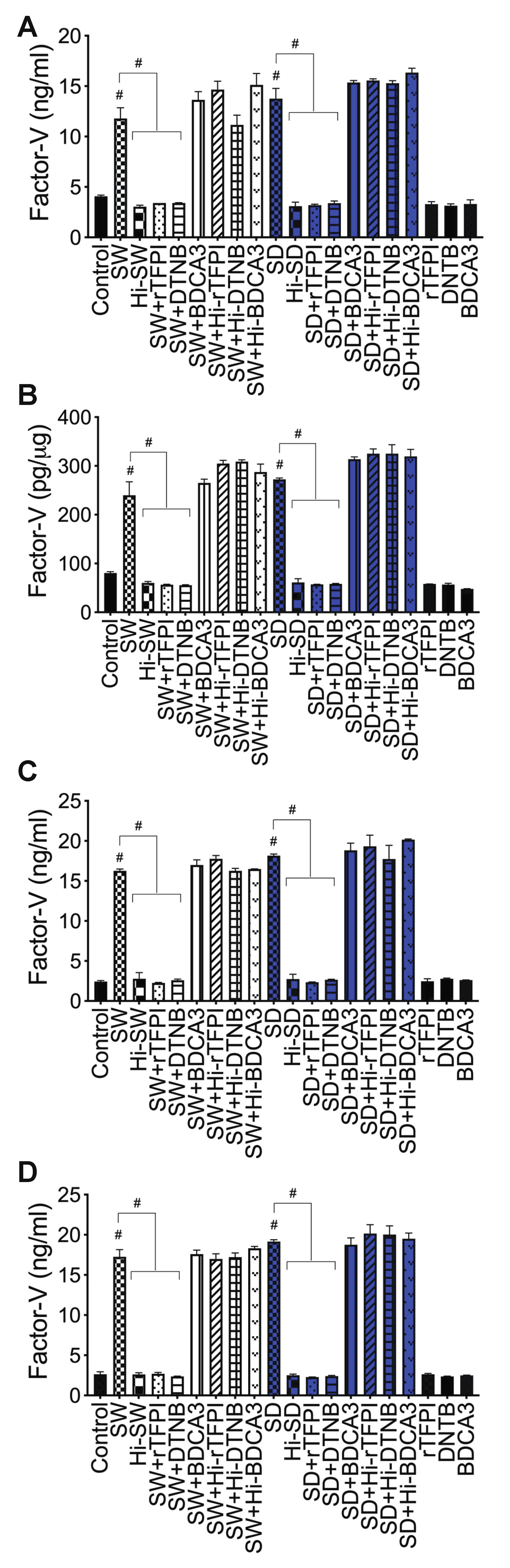
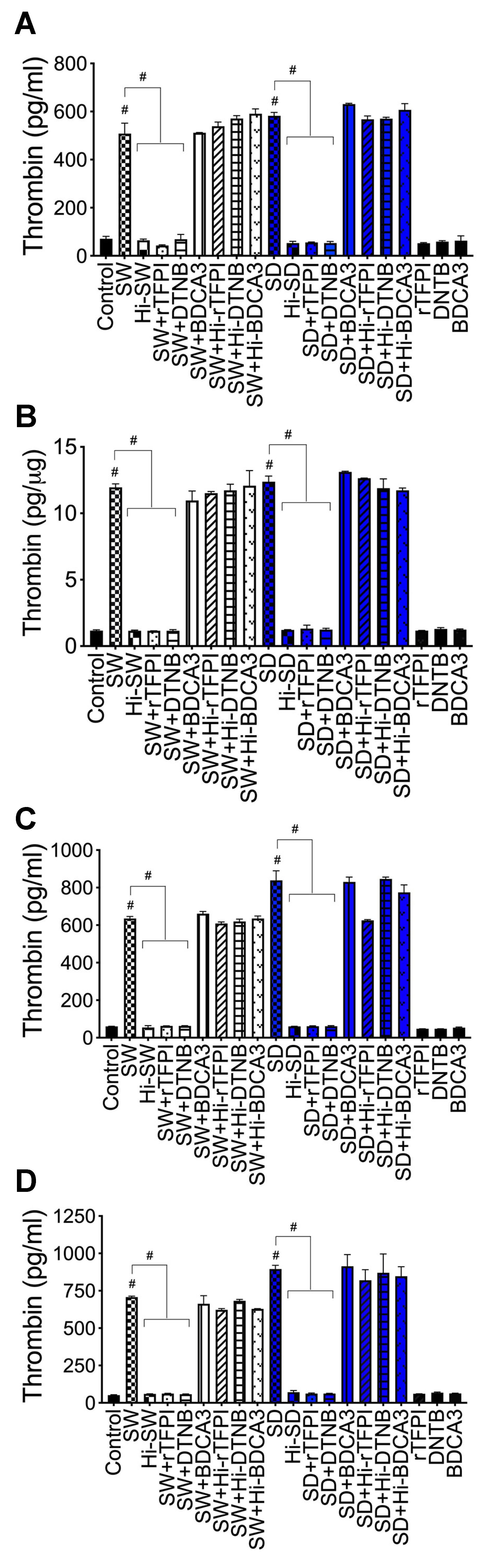
2.3. Exposure of HLMEC and Neutrophils to S-Proteins and Endothelial–Neutrophil Interactions Increased the Expression and Secretion of Factor-V and Thrombin
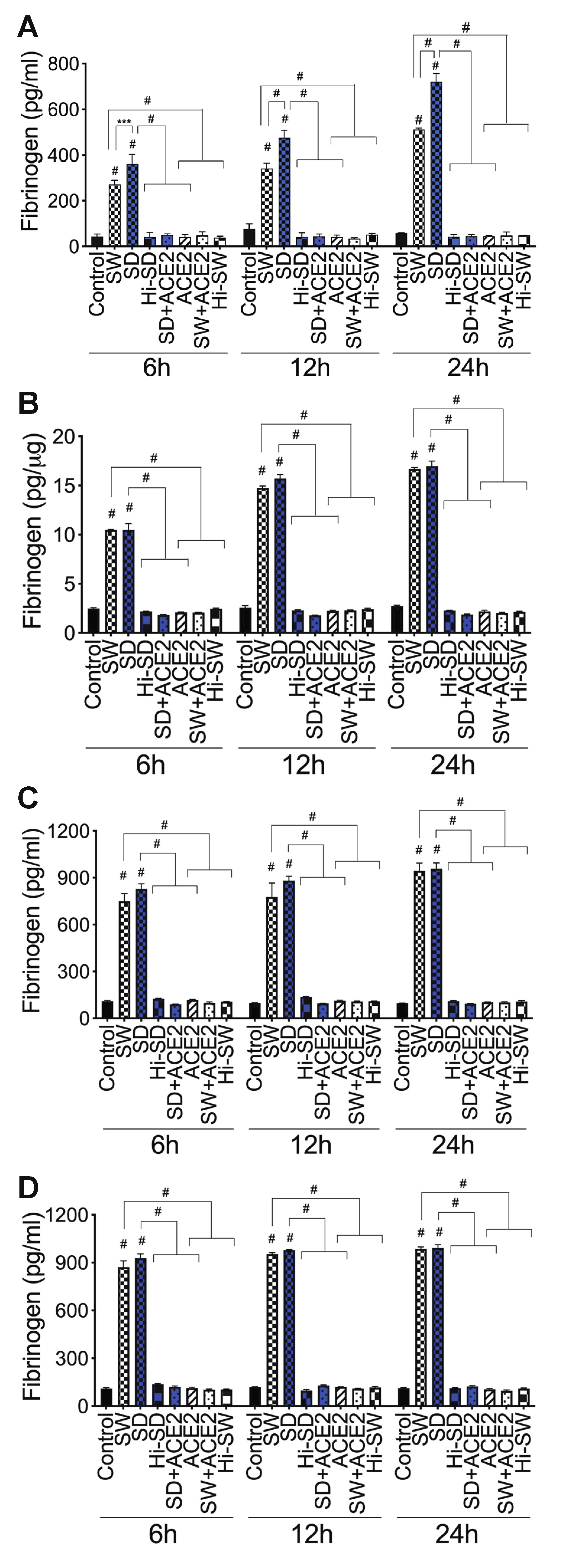
2.4. Exposure of HLMEC and Neutrophils to S-Proteins and Endothelial–Neutrophil Interactions Increased the Expression and Secretion of Fibrinogen
2.5. Exposure of HLMEC and Neutrophils to S-Proteins and Endothelial–Neutrophil Interactions Inhibits TFPI
2.6. rTFPI Blocked S-Proteins-Induced Expression and Secretion of Factor-V, Thrombin, and Fibrinogen
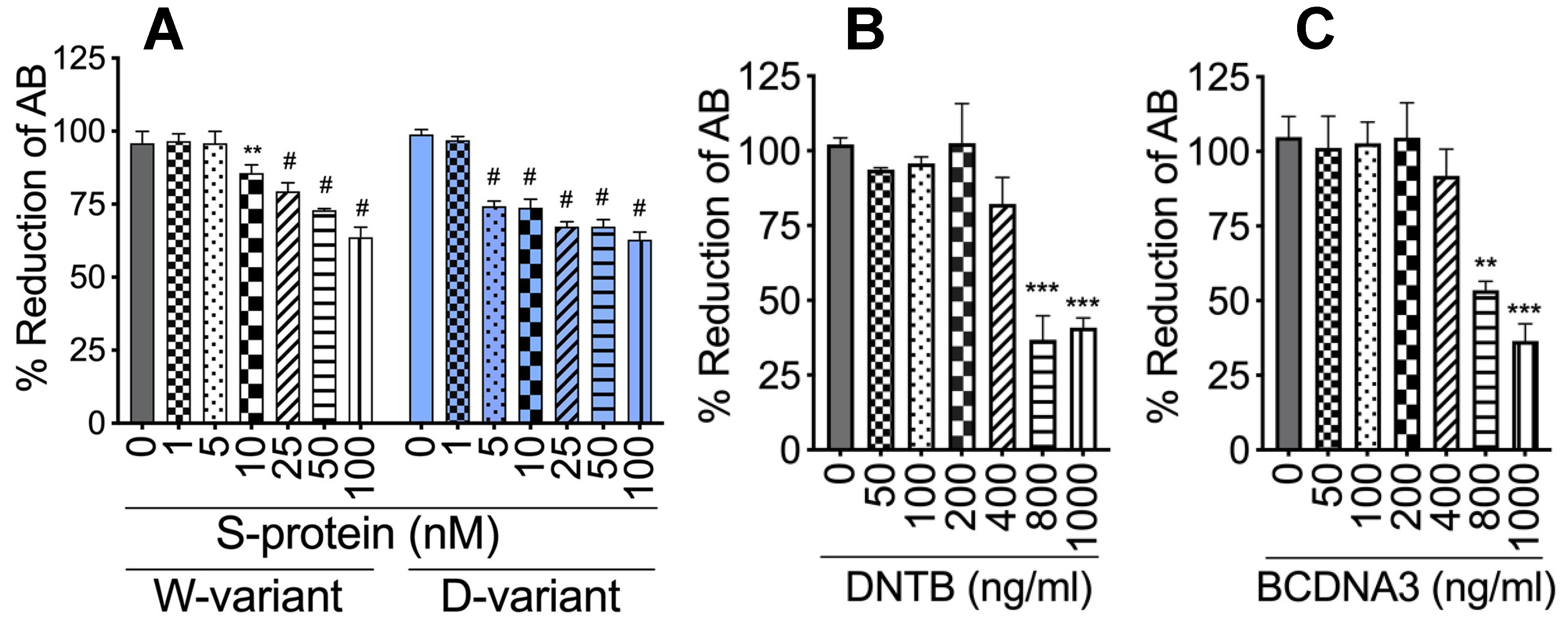
2.7. DTNB Blocked S-Proteins-Induced Expression and Secretion of Factor-V, Thrombin, and Fibrinogen
2.8. Thrombomodulin Blocked S-Protein-Induced Expression and Secretion of Fibrinogen but Had No Effect on S-Protein-Induced Expression and Secretion of Factor-V or Thrombin
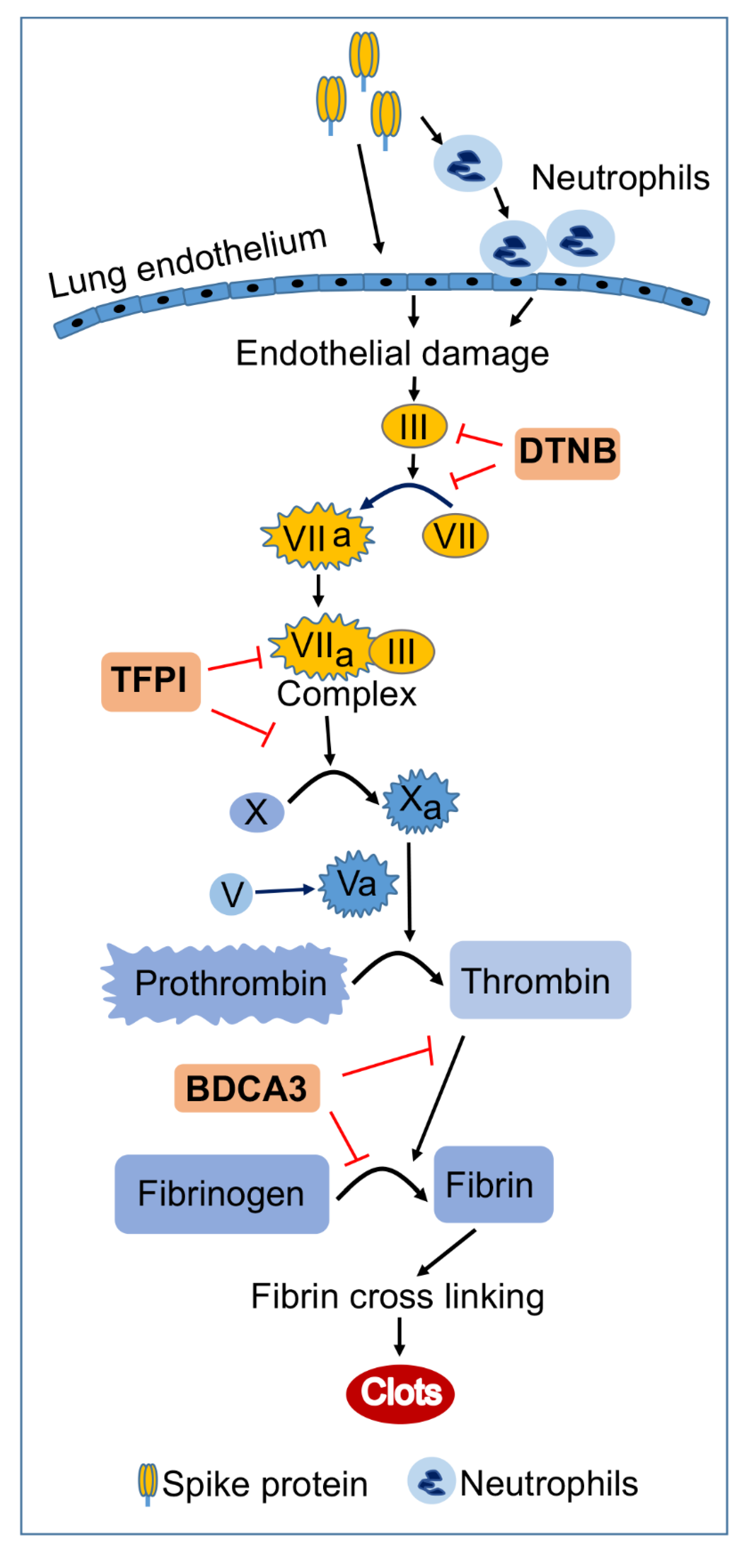
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cytotoxicity Assay
4.3. Primary Human Lung Microvascular Endothelial Cells and Neutrophils
4.4. Cells Treatment and Endothelial-Neutrophil Co-Culture
4.5. RNA Isolation and Real-Time PCR
4.6. Human Factor-V, Thrombin, and Fibrinogen ELISA
4.7. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DTNB | 5:5′-dithio-bis-(2-nitrobenzoic acid |
| TFPI | Tissue factor pathway inhibitor |
| rTFPI | Recombinant tissue factor pathway inhibitor |
| BDCA3/TM | Thrombomodulin |
| III/TF | Factor-III/tissue factor |
| VII | Factor-VII |
| VIIa | Activated factor-VII |
| X | Factor-X |
| Xa | Activated factor-X |
| V | Factor-V |
| Va | Activated factor-V |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus-2 |
| COVID-19 | Coronavirus disease 2019 |
| ACE2 | Angiotensin-converting enzyme-2 |
| rhACE2 | Recombinant human ACE2 |
| S-proteins | Spike proteins |
| SW | Spike protein: Wuhan variant |
| SD | Spike protein: Delta variant |
| Hi | Heat-inactivated |
| RBD | Receptor binding domain |
| PD | Protease domain |
| ELISA | Enzyme-linked immunosorbent assay |
| PCR | Polymerase chain reaction |
| cDNA | Complementary DNA |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| Cat. # | Catalog number |
References
- Mackenzie, J.S.; Smith, D.W. COVID-19: A novel zoonotic disease caused by a coronavirus from China: What we know and what we don’t. Microbiol. Aust. 2020, 41, 45. [Google Scholar] [CrossRef] [PubMed]
- Peeri, N.C.; Shrestha, N.; Rahman, M.S.; Zaki, R.; Tan, Z.; Bibi, S.; Baghbanzadeh, M.; Aghamohammadi, N.; Zhang, W.; Haque, U. The SARS, MERS and novel coronavirus (COVID-19) epidemics, the newest and biggest global health threats: What lessons have we learned? Int. J. Epidemiol. 2020, 49, 717–726. [Google Scholar] [CrossRef]
- JHU. Coronavirus Resource Center: COVID-19 in the USA. 2022. Available online: https://coronavirus.jhu.edu/ (accessed on 22 August 2022).
- CDC. United States COVID-19 Cases and Deaths by State. US Center for Disease Control and Prevention. 2022. Available online: https://www.cdc.gov/covid-data-tracker/#cases (accessed on 22 August 2022).
- WHO. Coronavirus disease (COVID-19) pandemic. World Health Organization. 2022. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 22 August 2022).
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; da Silva, L.F.F.; de Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020, 7, e553–e555. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Procko, E. The sequence of human ACE2 is suboptimal for binding the S spike protein of SARS coronavirus 2. bioRxiv 2020. [Google Scholar] [CrossRef]
- McKenna, E.; Wubben, R.; Isaza-Correa, J.M.; Melo, A.M.; Mhaonaigh, A.U.; Conlon, N.; O’Donnell, J.S.; Ni Cheallaigh, C.; Hurley, T.; Stevenson, N.J.; et al. Neutrophils in COVID-19: Not Innocent Bystanders. Front. Immunol. 2022, 13, 864387. [Google Scholar] [CrossRef] [PubMed]
- Calvert, B.A.; Quiroz, E.J.; Lorenzana, Z.; Doan, N.; Kim, S.; Senger, C.N.; Wallace, W.D.; Salomon, M.P.; Henley, J.; Ryan, A.L. Neutrophilic inflammation promotes SARS-CoV-2 infectivity and augments the inflammatory responses in airway epithelial cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Iizuka, K.; Kusunoki, A.; Machida, T.; Hirafuji, M. Angiotensin II reduces membranous angiotensin-converting enzyme 2 in pressurized human aortic endothelial cells. J. Renin Angiotensin Aldosterone Syst. 2009, 10, 210–215. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef]
- Perico, L.; Morigi, M.; Galbusera, M.; Pezzotta, A.; Gastoldi, S.; Imberti, B.; Perna, A.; Ruggenenti, P.; Donadelli, R.; Benigni, A.; et al. SARS-CoV-2 Spike Protein 1 Activates Microvascular Endothelial Cells and Complement System Leading to Platelet Aggregation. Front. Immunol. 2022, 13, 827146. [Google Scholar] [CrossRef]
- Avolio, E.; Carrabba, M.; Milligan, R.; Kavanagh Williamson, M.; Beltrami, A.P.; Gupta, K.; Elvers, K.T.; Gamez, M.; Foster, R.R.; Gillespie, K.; et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: A potential non-infective mechanism of COVID-19 microvascular disease. Clin. Sci. 2021, 135, 2667–2689. [Google Scholar] [CrossRef]
- Nuovo, G.J.; Magro, C.; Shaffer, T.; Awad, H.; Suster, D.; Mikhail, S.; He, B.; Michaille, J.J.; Liechty, B.; Tili, E. Endothelial cell damage is the central part of COVID-19 and a mouse model induced by injection of the S1 subunit of the spike protein. Ann. Diagn Pathol. 2021, 51, 151682. [Google Scholar] [CrossRef]
- Laurence, J.; Nuovo, G.; Racine-Brzostek, S.E.; Seshadri, M.; Elhadad, S.; Crowson, A.N.; Mulvey, J.J.; Harp, J.; Ahamed, J.; Magro, C. Premortem Skin Biopsy Assessing Microthrombi, Interferon Type I Antiviral and Regulatory Proteins, and Complement Deposition Correlates with Coronavirus Disease 2019 Clinical Stage. Am. J. Pathol. 2022, 192, 1282–1294. [Google Scholar] [CrossRef]
- George, S.; Pal, A.C.; Gagnon, J.; Timalsina, S.; Singh, P.; Vydyam, P.; Munshi, M.; Chiu, J.E.; Renard, I.; Harden, C.A.; et al. Evidence for SARS-CoV-2 Spike Protein in the Urine of COVID-19 Patients. Kidney360 2021, 2, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C. Could SARS-CoV-2 Spike Protein Be Responsible for Long-COVID Syndrome? Mol. Neurobiol. 2022, 59, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, J.; Laurence, J. Long COVID endotheliopathy: Hypothesized mechanisms and potential therapeutic approaches. J. Clin. Investig. 2022, 132, e161167. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. The molecular basis of blood coagulation. Cell 1988, 53, 505–518. [Google Scholar] [CrossRef]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arter. Thromb Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Baugh, R.J.; Broze, G.J., Jr.; Krishnaswamy, S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J. Biol. Chem. 1998, 273, 4378–4386. [Google Scholar] [CrossRef]
- Bajaj, M.S.; Birktoft, J.J.; Steer, S.A.; Bajaj, S.P. Structure and biology of tissue factor pathway inhibitor. Thromb. Haemost. 2001, 86, 959–972. [Google Scholar]
- Lwaleed, B.A.; Bass, P.S. Tissue factor pathway inhibitor: Structure, biology and involvement in disease. J. Pathol. 2006, 208, 327–339. [Google Scholar] [CrossRef]
- Maroney, S.A.; Hansen, K.G.; Mast, A.E. Cellular expression and biological activities of alternatively spliced forms of tissue factor pathway inhibitor. Curr. Opin. Hematol. 2013, 20, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, K.; Tasaki, T.; Yamauchi, T.; Iwasaki, H.; Ueda, T. Successful Administration of Recombinant Human Soluble Thrombomodulin alpha (Recomodulin) for Disseminated Intravascular Coagulation during Induction Chemotherapy in an Elderly Patient with Acute Monoblastic Leukemia Involving the t(9;11)(p22;q23) MLL/AF9 Translocation. Case Rep. Hematol. 2011, 2011, 273070. [Google Scholar] [PubMed]
- Loghmani, H.; Conway, E.M. Exploring traditional and nontraditional roles for thrombomodulin. Blood 2018, 132, 148–158. [Google Scholar] [CrossRef]
- Greenberg, C.S.; Miraglia, C.C.; Rickles, F.R.; Shuman, M.A. Cleavage of blood coagulation factor XIII and fibrinogen by thrombin during in vitro clotting. J. Clin. Investig. 1985, 75, 1463–1470. [Google Scholar] [CrossRef]
- Lane, D.A.; Philippou, H.; Huntington, J.A. Directing thrombin. Blood 2005, 106, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Subrahmanian, S.; Borczuk, A.; Salvatore, S.; Fung, K.M.; Merrill, J.T.; Laurence, J.; Ahamed, J. Tissue factor upregulation is associated with SARS-CoV-2 in the lungs of COVID-19 patients. J. Thromb. Haemost. 2021, 19, 2268–2274. [Google Scholar] [CrossRef]
- Guervilly, C.; Bonifay, A.; Burtey, S.; Sabatier, F.; Cauchois, R.; Abdili, E.; Arnaud, L.; Lano, G.; Pietri, L.; Robert, T.; et al. Dissemination of extreme levels of extracellular vesicles: Tissue factor activity in patients with severe COVID-19. Blood Adv. 2021, 5, 628–634. [Google Scholar] [CrossRef]
- Canas, C.A.; Canas, F.; Bautista-Vargas, M.; Bonilla-Abadia, F. Role of Tissue Factor in the Pathogenesis of COVID-19 and the Possible Ways to Inhibit It. Clin. Appl. Thromb. Hemost. 2021, 27, 10760296211003983. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler Thromb Vasc Biol 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, W.R.M. Regulation of tissue factor expression. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6620/ (accessed on 20 July 2022).
- Osterud, B.; Bjorklid, E. Tissue factor in blood cells and endothelial cells. Front. Biosci. 2012, 4, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Duan, Y.; Bao, T.; Gu, J.; Chen, Y.; Li, Y.; Mao, S.; Chen, Y.; Xie, W. The values of coagulation function in COVID-19 patients. PLoS ONE 2020, 15, e0241329. [Google Scholar] [CrossRef]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.B. Thromboinflammation in COVID-19 acute lung injury. Paediatr. Respir. Rev. 2020, 35, 20–24. [Google Scholar] [CrossRef]
- Matafonov, A.; Sarilla, S.; Sun, M.F.; Sheehan, J.P.; Serebrov, V.; Verhamme, I.M.; Gailani, D. Activation of factor XI by products of prothrombin activation. Blood 2011, 118, 437–445. [Google Scholar] [CrossRef]
- Huntington, J.A. Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 2005, 3, 1861–1872. [Google Scholar] [CrossRef]
- Coughlin, S.R. How the protease thrombin talks to cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11023–11027. [Google Scholar] [CrossRef]
- Dubey, A.; Choudhary, S.; Kumar, P.; Tomar, S. Emerging SARS-CoV-2 Variants: Genetic Variability and Clinical Implications. Curr. Microbiol. 2021, 79, 20. [Google Scholar] [CrossRef]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L.; Geneva Centre for Emerging Viral Diseases. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef] [PubMed]
- Koelle, K.; Martin, M.A.; Antia, R.; Lopman, B.; Dean, N.E. The changing epidemiology of SARS-CoV-2. Science 2022, 375, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Broze, G.J., Jr.; Girard, T.J. Tissue factor pathway inhibitor: Structure-function. Front. Biosci. 2012, 17, 262–280. [Google Scholar] [CrossRef]
- Ellery, P.E.; Adams, M.J. Tissue factor pathway inhibitor: Then and now. Semin. Thromb. Hemost. 2014, 40, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E. Tissue Factor Pathway Inhibitor: Multiple Anticoagulant Activities for a Single Protein. Arter. Thromb. Vasc. Biol. 2016, 36, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta 2014, 1840, 838–846. [Google Scholar] [CrossRef]
- Chen, V.M. Tissue factor de-encryption, thrombus formation, and thiol-disulfide exchange. Semin. Thromb. Hemost. 2013, 39, 40–47. [Google Scholar] [CrossRef]
- Chen, V.M.; Ahamed, J.; Versteeg, H.H.; Berndt, M.C.; Ruf, W.; Hogg, P.J. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry 2006, 45, 12020–12028. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Banerjee, S.; Sen, P. Contribution of allosteric disulfide in the structural regulation of membrane-bound tissue factor-factor VIIa binary complex. J. Biomol. Struct. Dyn. 2019, 37, 3707–3720. [Google Scholar] [CrossRef]
- Gallagher, T.M. Murine coronavirus membrane fusion is blocked by modification of thiols buried within the spike protein. J. Virol. 1996, 70, 4683–4690. [Google Scholar] [CrossRef]
- Ryser, H.J.; Levy, E.M.; Mandel, R.; DiSciullo, G.J. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci. USA 1994, 91, 4559–4563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, M.; Ekstrom, M.; Garoff, H. Isomerization of the intersubunit disulphide-bond in Env controls retrovirus fusion. EMBO J. 2004, 23, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Hati, S.; Bhattacharyya, S. Impact of Thiol-Disulfide Balance on the Binding of Covid-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor. ACS Omega 2020, 5, 16292–16298. [Google Scholar] [CrossRef] [PubMed]
- Stantchev, T.S.; Paciga, M.; Lankford, C.R.; Schwartzkopff, F.; Broder, C.C.; Clouse, K.A. Cell-type specific requirements for thiol/disulfide exchange during HIV-1 entry and infection. Retrovirology 2012, 9, 97. [Google Scholar] [CrossRef]
- Mirazimi, A.; Mousavi-Jazi, M.; Sundqvist, V.A.; Svensson, L. Free thiol groups are essential for infectivity of human cytomegalovirus. J. Gen. Virol. 1999, 80 Pt 11 Pt 11, 2861–2865. [Google Scholar] [CrossRef]
- Khanna, K.; Raymond, W.; Jin, J.; Charbit, A.R.; Gitlin, I.; Tang, M.; Werts, A.D.; Barrett, E.G.; Cox, J.M.; Birch, S.M.; et al. Thiol drugs decrease SARS-CoV-2 lung injury in vivo and disrupt SARS-CoV-2 spike complex binding to ACE2 in vitro. bioRxiv 2021. [Google Scholar] [CrossRef]
- Khanna, K.; Raymond, W.; Jin, J.; Charbit, A.R.; Gitlin, I.; Tang, M.; Werts, A.D.; Barrett, E.G.; Cox, J.M.; Birch, S.M.; et al. Exploring antiviral and anti-inflammatory effects of thiol drugs in COVID-19. Am. J. Physiology. Lung Cell. Mol. Physiol. 2022, 323, L372–L389. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.L.; Soriano, J.B.; Gonzalez, Y.; Lumbreras, S.; Ancochea, J.; Echeverry, C.; Rodriguez, J.M. Use of N-Acetylcysteine at high doses as an oral treatment for patients hospitalized with COVID-19. Sci. Prog. 2022, 105, 368504221074574. [Google Scholar] [CrossRef]
- Cui, X.Y.; Tjonnfjord, G.E.; Kanse, S.M.; Dahm, A.E.A.; Iversen, N.; Myklebust, C.F.; Sun, L.; Jiang, Z.X.; Ueland, T.; Campbell, J.J.; et al. Tissue factor pathway inhibitor upregulates CXCR7 expression and enhances CXCL12-mediated migration in chronic lymphocytic leukemia. Sci. Rep. 2021, 11, 5127. [Google Scholar] [CrossRef]
- Kanmogne, G.D.; Kennedy, R.C.; Grammas, P. Analysis of human lung endothelial cells for susceptibility to HIV type 1 infection, coreceptor expression, and cytotoxicity of gp120 protein. AIDS Res. Hum. Retrovir. 2001, 17, 45–53. [Google Scholar] [CrossRef]
- Kanmogne, G.D.; Primeaux, C.; Grammas, P. Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem. Biophys. Res. Commun. 2005, 333, 1107–1115. [Google Scholar] [CrossRef]
- Li, H.; Singh, S.; Potula, R.; Persidsky, Y.; Kanmogne, G.D. Dysregulation of claudin-5 in HIV-induced interstitial pneumonitis and lung vascular injury. Protective role of peroxisome proliferator-activated receptor-gamma. Am. J. Respir. Crit. Care Med. 2014, 190, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Woollard, S.M.; Li, H.; Singh, S.; Yu, F.; Kanmogne, G.D. HIV-1 induces cytoskeletal alterations and Rac1 activation during monocyte-blood-brain barrier interactions: Modulatory role of CCR5. Retrovirology 2014, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Yang, B.; Gendelman, H.E.; Persidsky, Y.; Kanmogne, G.D. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008, 111, 2062–2072. [Google Scholar] [CrossRef] [PubMed]
- Bhargavan, B.; Kanmogne, G.D. Toll-Like Receptor-3 Mediates HIV-1-Induced Interleukin-6 Expression in the Human Brain Endothelium via TAK1 and JNK Pathways: Implications for Viral Neuropathogenesis. Mol. Neurobiol. 2018, 55, 5976–5992. [Google Scholar] [CrossRef] [PubMed]
- Bhargavan, B.; Woollard, S.M.; McMillan, J.E.; Kanmogne, G.D. CCR5 antagonist reduces HIV-induced amyloidogenesis, tau pathology, neurodegeneration, and blood-brain barrier alterations in HIV-infected hu-PBL-NSG mice. Mol. Neurodegener 2021, 16, 78. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhargavan, B.; Kanmogne, G.D. SARS-CoV-2 Spike Proteins and Cell–Cell Communication Inhibits TFPI and Induces Thrombogenic Factors in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for COVID-19 Coagulopathy Pathogenesis. Int. J. Mol. Sci. 2022, 23, 10436. https://doi.org/10.3390/ijms231810436
Bhargavan B, Kanmogne GD. SARS-CoV-2 Spike Proteins and Cell–Cell Communication Inhibits TFPI and Induces Thrombogenic Factors in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for COVID-19 Coagulopathy Pathogenesis. International Journal of Molecular Sciences. 2022; 23(18):10436. https://doi.org/10.3390/ijms231810436
Chicago/Turabian StyleBhargavan, Biju, and Georgette D. Kanmogne. 2022. "SARS-CoV-2 Spike Proteins and Cell–Cell Communication Inhibits TFPI and Induces Thrombogenic Factors in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for COVID-19 Coagulopathy Pathogenesis" International Journal of Molecular Sciences 23, no. 18: 10436. https://doi.org/10.3390/ijms231810436
APA StyleBhargavan, B., & Kanmogne, G. D. (2022). SARS-CoV-2 Spike Proteins and Cell–Cell Communication Inhibits TFPI and Induces Thrombogenic Factors in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for COVID-19 Coagulopathy Pathogenesis. International Journal of Molecular Sciences, 23(18), 10436. https://doi.org/10.3390/ijms231810436