1. Introduction
Telomeres are nucleoprotein structures positioned at the natural ends of linear eukaryotic chromosomes, where they avoid unwanted activation of DNA repair machineries, telomeric DNA degradation and telomeric fusions [
1,
2]. Mammalian telomeres contain the multiprotein complex shelterin, which assembles through protein-DNA and protein-protein interactions [
1,
2]. In addition, telomeres contain the long noncoding RNA TERRA, which is transcribed from subtelomeric promoters towards the end of chromosomes by RNA polymerase II. Individual TERRA molecules comprise unique subtelomeric RNA sequences followed by (UUAGGG)n tracts of variable length [
3,
4,
5]. A fraction of human TERRA associates with telomeric chromatin through RNA:DNA hybrids (telomeric R-loops or telR-loops) forming between the UUAGGG repeats and the template, C-rich telomeric DNA strand [
3,
6,
7].
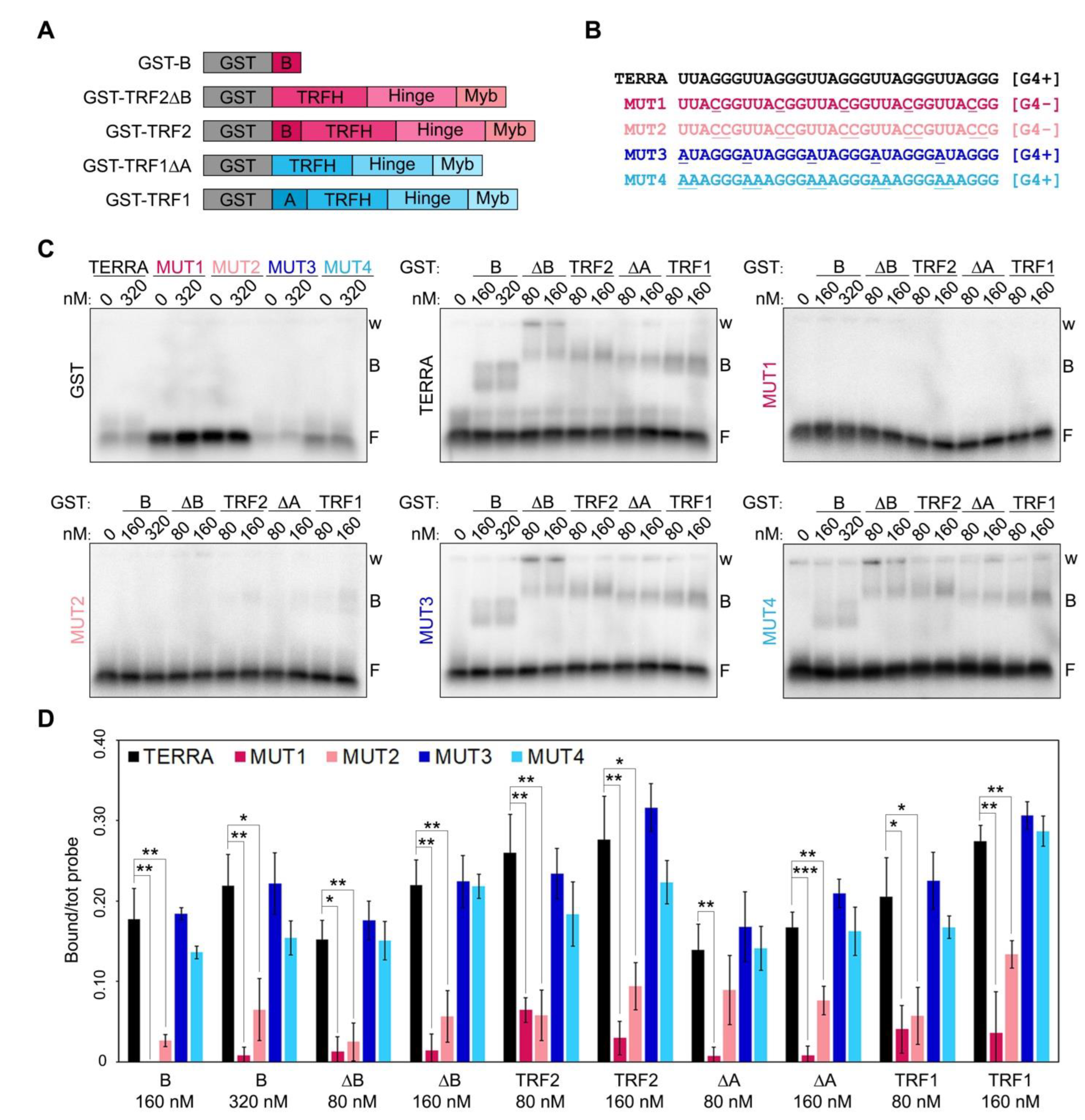
Several laboratories including ours demonstrated that the two human shelterin proteins TRF1 and TRF2 establish an intricate and functionally relevant crosstalk with TERRA. Recombinant TRF2 binds to TERRA oligonucleotides in vitro through different domains including its N-terminal glycine-arginine-rich (GAR) domain, a 44 amino acid long peptide also dubbed B domain due to its basic nature [
2,
7,
8,
9,
10,
11]. Recombinant TRF1 also binds to TERRA, through interactions occurring outside of its N-terminal, 66 amino acid long acidic (A) domain [
7,
10]. Confirming the validity of these in vitro observations, mammalian TRF1 and TRF2 physically associate with TERRA (UUAGGG)n sequences in human and mouse cells [
10,
12,
13,
14].
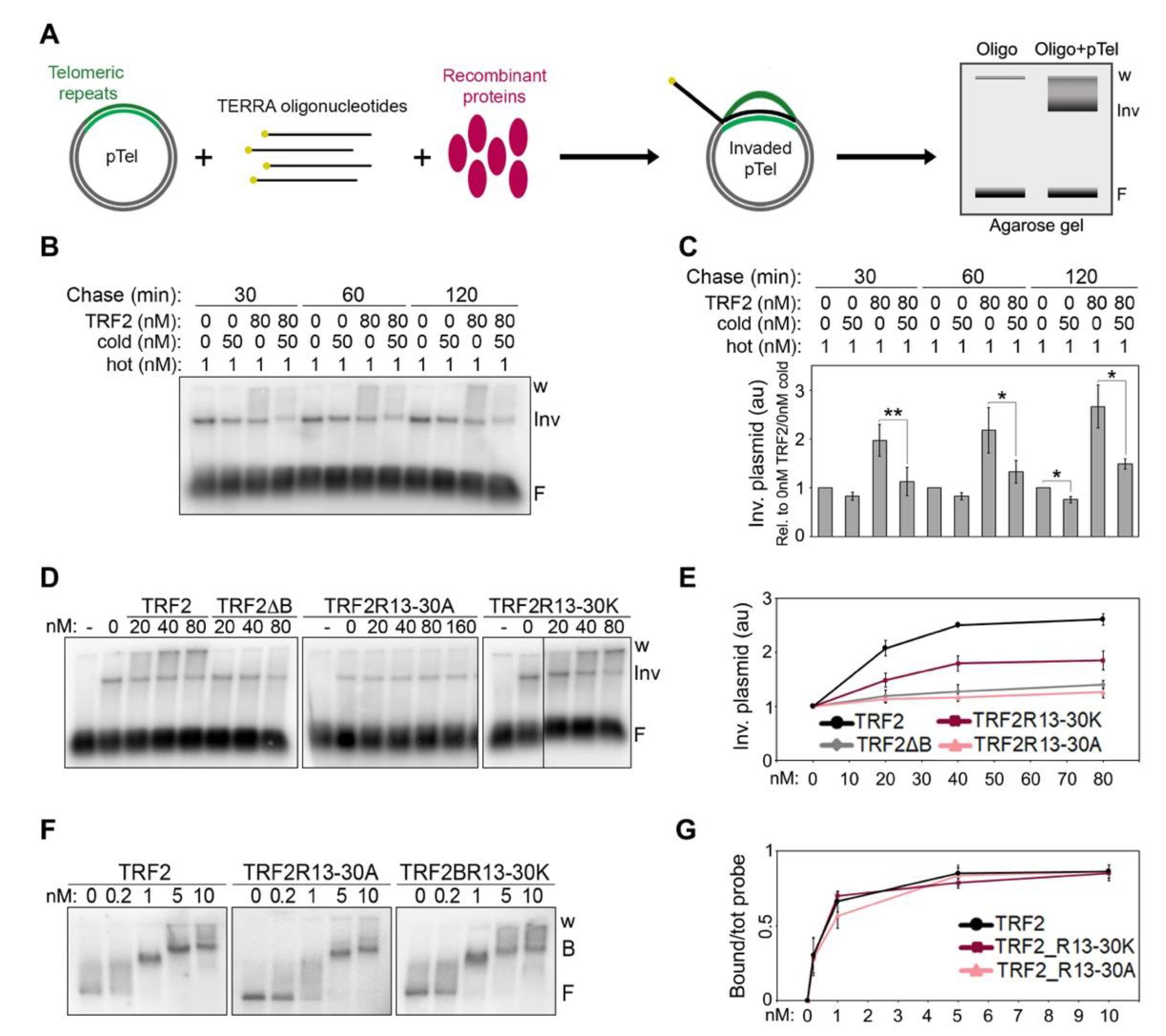
We showed that TRF2 stimulates TERRA invasion of telomeric double-stranded (ds) DNA in vitro, leading to the formation of telR-loops; this activity relies on TRF2 B domain, as a TRF2 variant lacking the B domain (TRF2ΔB) is unable to stimulate TERRA invasion [
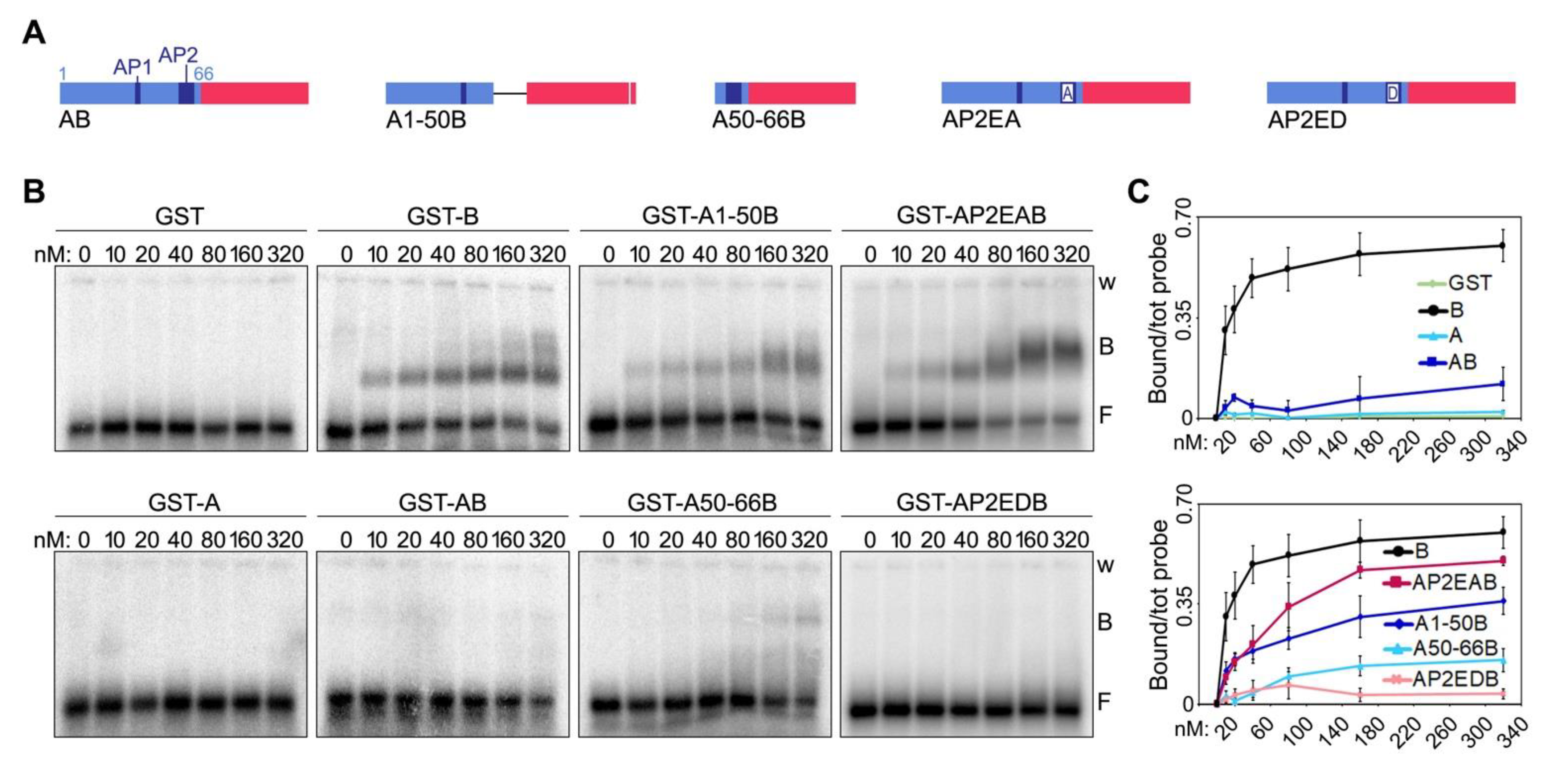
7]. Conversely, TRF1 A domain, while not being able to bind to TERRA, inhibits TERRA binding to TRF2 B domain and suppresses TRF2 ability to stimulate TERRA invasion [
7]. In cells, TRF2 over-expression or replacement of TRF1 with a variant lacking the A domain (TRF1ΔA) causes aberrant accumulation of telR-loops which, in turn, provoke telomeric replication stress and rapid telomere loss [
7]. While the functions associated to TRF2-induced telR-loops are still being studied, our data allowed us to propose that those telR-loops must be kept in check by TRF1 in order to avoid telomere instability.
Despite the importance of the complex interplay between TERRA, TRF proteins and telR-loops, the molecular and structural details of these interactions are still very limited. The UUAGGG tracts of TERRA can form G-quadruplexes (G4s) containing stacked Hoogsteen-bonded G-quartet motifs stabilized by monovalent cations including K+ [
9,
15]. Recombinant full-length TRF2 and its B domain alone bind to TERRA in a G4-dependent manner, as those interactions are stabilized by G4 stabilizers and, conversely, are lost when nucleotide substitutions impairing G4 formation are introduced in the RNA substrates [
9,
11,
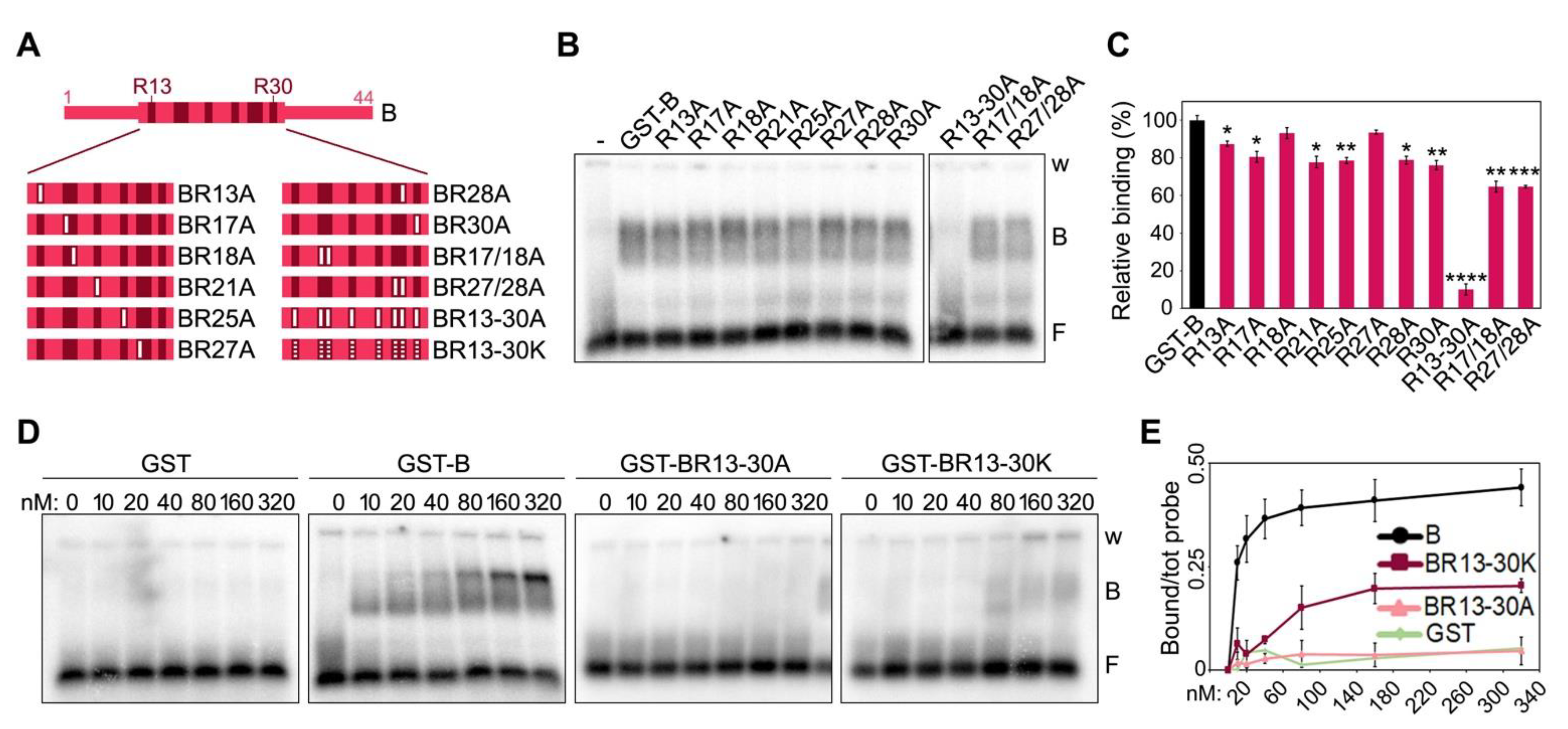
15]. Moreover, the substitution of 10 arginines (Rs) within the B domain into alanines (As) abolished the binding to TERRA, highlighting the importance of the GAR motif organization for RNA G4 binding [
11]. To expand our understanding of the crosstalk between TERRA and TRF proteins, we set up an extensive array of biochemical assays using full-length and deleted TRF proteins or amino acid substitution variants in combination with different RNA oligonucleotides.
3. Discussion
The functional relevance of TERRA interactions with human TRF2 B domain has already been clarified, as these interactions promote the formation of telR-loops [
7]. It remains to be understood when those TRF2-stimulated telR-loops are needed; it is possible that they become essential when telomeres shorten and produce more TERRA, in order to induce telomere elongation through homology-directed repair, as it has been proposed for budding and fission yeasts [
17,
18,
19,
20]. The relevance of the binding of other domains of TRF1 and TRF2 to TERRA still needs to be explored; however, they might also support telomere stability for example by regulating shelterin assembly. We have demonstrated here that both TRF1 and TRF2 binding to TERRA UUAGGG repeats depends on the ability of the latter to form G4s. In this light, our data strengthen the notion that G4 structures play crucial roles in safeguarding genome integrity [
21]. Interestingly, it was recently demonstrated that the DNA repair factor RAD51 also binds to TERRA and promotes the formation of telR-loops in vitro [
22]. It is thus possible that TRF2 and RAD51 cooperate in regulating telR-loop formation, perhaps by associating with the same TERRA moieties. Biochemical assays similar to the ones employed here should help probe this assumption, for example, by determining whether RAD51 binding to TERRA also depends on G4 structures.
The interaction of TRF2 B domain with TERRA G4s was reported also by the Lieberman laboratory [
11]. In the same report, the authors showed that the small molecule N-methyl mesoporphyrin IX (NMM) binds with relatively high affinity to TERRA G4s and disrupts the TERRA-TRF2 B domain complex. Moreover, when they treated cells with NMM, they observed features of compromised telomere stability, including DNA damage marks at telomeres, rapid telomere shortening and under-replicated fragile telomeres [
11]. While all those features were ascribed to the disruption of TERRA-B domain complexes, it is also possible that some of them could derive from the disruption of other interactions between TERRA and TRF2 regions outside of the B domain or TRF1. It will be important to identify all TERRA binding sites present in both TRF1 and TRF2 and study the contribution of each of them towards telomere stability.
We have also defined, with unprecedented resolution, the molecular details of how TRF2 B domain binds to TERRA and TRF1 A domain inhibits this binding. As for TRF2 B domain, the 8 Rs clustered within the GAR domain are necessary for binding to TERRA. Replacing them with positively charged Ks (BR13-30K mutant) impairs TERRA binding, although not completely, and the corresponding TRF2 full-length protein (TRF2R13-30K mutant) is still partly able to induce telR-loop formation. This suggests that electrostatic interactions and other types of contacts, likely established through arginine side chains, support TERRA-B domain interactions and consequent telR-loop formation. Consistently, it has been shown that the multivalent guanidinium group of arginines establishes multiple interactions with the backbone phosphates of all RNA bases, thereby stabilizing RNA-protein complexes [
23]. In contrast, the inhibitory activity associated to TRF1 A domain appears to largely, if not exclusively, depend on the charges associated to the eight Es clustered within the AP2; indeed, the negatively charged AP2EDB mutant inhibits TERRA-B domain interactions as efficiently as the original AB protein. Overall, it is evident that the contribution of charged amino acids is fundamental in establishing the regulated interplay between TERRA and TRF proteins. Given the negatively charged nature of TERRA RNA, we speculate that positively charged Rs attract TERRA to the B domain and negatively charged Es repel it from telomeres or shield TRF2 B domain through RNA mimicry [
24]. Our model also implies that independent, negatively charged patches placed in proximity of TRF2 at telomeres could exert functions similar to the ones exerted by TRF1 A domain. Interestingly, human RAP1, another shelterin component that directly interacts with TRF2, also contains an acidic domain comprised between amino acids 214 and 304 [
2] ending with a stretch of eight consecutive Es. Because this acidic domain is located immediately before the interface interacting with TRF2, it is highly possible that RAP1 also regulates TERRA-TRF2 complex and, in turn, telR-loop formation.
TERRA-TRF protein interactions, by regulating telR-loop formation and telomere stability, might protect cellular and tissue homeostasis and avoid disease conditions. A survey of cancer-associated mutations found in TRF1 and TRF2 at the cBioPortal for Cancer Genomics [
25,
26] reveals that none of them is located within TRF2 B domain, suggesting a strong counterselection against altered B domain variants. Conversely, several mutations leading to amino acid substitutions within TRF1 A domain can be identified. One of those mutations causes a E to K substitution within the AP1 (E36K, found in one lung adenocarcinoma and two cutaneous melanomas) and two of them cause E to K substitutions within the AP2 (E55K and E58K, both found in bladder urothelial carcinomas). The impact of these substitutions on the inhibitory activity of TRF1 A domain still needs to be tested; nonetheless, it is tempting to speculate that an impaired inhibitory activity might cause telomere instability and contribute to cancer etiology and/or development.
Finally, we show that TERRA-TRF2 B domain interactions are conserved in mouse, while the regulatory function of TRF1 A domain is not. This suggests that mouse TRF2 promotes telR-loop formation, while mouse TRF1 cannot suppress them. If that were the case, factors other than TRF1, RAP1, for example, could be involved in restricting TRF2-stimulated telR-loops. However, conditional deletion of TRF1 in mouse embryonic fibroblasts leads to the appearance of telomere free chromosome ends [
27], a phenotype also induced by TRF1 depletion in human cells [
7]. We previously showed that, in human cells, this defect is specifically averted by the A domain of TRF1 and derives from aberrant telR-loops, as it can be suppressed by over-expression of the RNA:DNA hybrid-specific nuclease RNaseH1 [
7]. While it is not known whether the telomere free ends induced by deletion of mouse TRF1 are also an outcome of aberrant telR-loops, it is possible that mouse TRF1 has retained the ability to suppress TRF2-induced telR-loops through mechanisms different from the ones in human. Further studies will be necessary to clarify the evolutionary conservation of the interactions between TERRA and TRF proteins and their regulation.
4. Materials and Methods
4.1. Plasmids
Plasmids pGEX-4T1 (GE Healthcare, Chicago, IL, USA) containing cDNAs of the proteins of interest N-terminally fused in frame to a Glutathione S-transferase (GST) sequence were used for expression in bacteria cells. Plasmids encoding for TRF proteins, TRF1ΔA, TRF2ΔB, the wild-type A and B domains, as well as the fusion of the two domains, were previously generated in the laboratory [
7]. All subsequent deletions and single or double amino acid substitutions were obtained using the Q5 site-directed mutagenesis kit (New England Biolabs, Ipswich, MA, USA). cDNAs for TRF2 variants R13-30K and R13-30A were synthesized at GeneScript (Piscataway, NJ, USA), and those for BL, mB, mBL, mA, AP2EAB and AP2EDB at Integrated DNA Technologies (IDT, Leuven, Belgium). These sequences were then cloned into the pGEX-4T1 vector. All plasmids generated were validated by sequencing. The pTel plasmid used in plasmid invasion assays was previously described [
7].
4.2. GST Protein Purification
The pGEX-4T1 plasmids were transformed into competent BL21 cells. Cells were grown in 5 mL LB media overnight at 37 °C and then 250 µL of those cultures were inoculated into 50 mL of fresh LB media. Cells were grown at 37 °C for 2 h, after which protein expression was induced with 50 μM IPTG (Sigma Aldrich, Merck Inc., Darmstad, Germany) for 5 h at 30 °C. Cells were collected by centrifugation (4200× g for 15 min at 4 °C) and lysed by sonication in 20 mL of GST pulldown buffer (50 mM potassium phosphate buffer pH 7, 1% Triton X-100, 100 mM NaCl, 5 mM EDTA, 0.15 mM PMSF) for proteins with molecular weights (MW) up to 50 kDa or in in Protein lysis buffer (50 mM Tris pH 8, 600 mM NaCl, 10% Glycerol, 1% Tween-20, 5 mM β-Mercaptoethanol, 1 mM PMSF) for proteins with MW above 50 kDa. Sonication was performed using a MSE Soniprep 150 apparatus (Sanyo, Osaka, Japan); samples were subjected to 2 cycles of 3 min “ON”, at maximum power, and 2 min “OFF”, while being kept on ice. Sonicated samples were then centrifuged at 4200× g for 20 min at 4 °C. After centrifugation, lysates were incubated with Glutathione agarose beads (Sigma Aldrich, Merck Inc., Darmstad, Germany) overnight at 4 °C on a tube roller. Beads were then washed three times with 5 mL of GST pulldown buffer and once with 2 mL of GST pulldown buffer low Triton (50 mM potassium phosphate buffer pH 7, 0.1% Triton X-100, 100 mM NaCl, 5 mM EDTA). Bound proteins were eluted in 200 μL of GST elution buffer (50 mM potassium phosphate buffer pH 7, 5 mM EDTA, 100 mM NaCl, 5% glycerol, 25 mM glutathione) for 20 min at 4 °C. Protein concentration and purity were determined using the Bradford protein assay (BioRad laboratories Inc., Hercules, CA, USA) and BSA reference samples, followed by fractionation in polyacrylamide gels and staining with BlueSafe reagent (NZYTech, Lisbon, Portugal). Note that all protein concentrations refer to monomers.
4.3. Electrophoretic Mobility Shift Assays (EMSAs)
For RNA EMSAs, 5-repeats oligonucleotides were synthesized at IDT (Leuven, Belgium). Sequences were as follow: TERRA, 5′-(UUAGGG)5-3′, MUT1, 5′-(UUACGG)5-3′, MUT2, 5′-(UUACCG)5-3′, MUT3, 5′-(AUAGGG)5-3′, MUT4, 5′-(AAAGGG)5-3′. Additionally, 12-repeat TERRA oligonucleotide [5′-(UUAGGG)12-3′] was obtained by in vitro transcription of a plasmid containing the respective DNA sequence downstream of a T7 promoter, using the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs, Ipswich, MA, USA), following the manufacturer’s instructions. For dsDNA EMSAs, a ds(TTAGGG) fragment was excised by restriction digestion from the pTel plasmid and contained approximately 1200 bp of telomeric repeats flanked by non-telomeric sequences of 48 (upstream) and 12 bp (downstream). RNA oligonucleotides and dsDNA fragments were 5′-end labeled with T4 polynucleotide kinase (New England Biolabs, Ipswich, MA, USA) and [γ32P]-ATP and then purified using the Oligo Clean and Concentrator Kit (Zymo Research, Irvine, CA, USA). For RNA EMSAs, recombinant proteins and oligos were incubated in 20 µL of EMSA buffer (50 mM HEPES pH 8, 1 mM DTT, 100 mM NaCl, 0.01% BSA, 2% glycerol) containing 50 ng/µL E. coli tRNAs (Sigma-Aldrich, Merck Inc., Darmstad, Germany) for 20 min on ice followed by 10 min at 25 °C. For dsDNA EMSAs, E. coli tRNAs were excluded from the reaction mix and incubation was carried out for 30 min at 25 °C. After incubation, 4 µL of 6× gel-loading buffer (30% glycerol, 0.3% bromophenol blue) were added to the reactions, which were then electrophoresed in agarose gels and pre-cooled 0.5× TBE. 2% agarose gels were used for EMSAs with 5-repeat RNA oligonucleotides, 1.5% for 12-repeat RNA oligonucleotides and 1% for dsDNA EMSAs. Gels were run at 70 V for 1 h at 4 °C and then dried and exposed to a phosphoimager screen. Radioactive signals were detected with a Typhoon FLA 9000 or an Amersham Typhoon IP imager (GE Healthcare, Chicago, IL, USA). ImageJ was used to quantify signals. The number of independent replicates (n) is indicated in figure legends.
4.4. Plasmid Invasion Assays
Plasmid invasion assays were performed adapting the protocol previously established in the laboratory [
7]. pTel plasmid was purified using the NZY Miniprep Kit (NZYTech, Lisbon, Portugal) and re-purified through Phenol-Chloroform extraction to assure removal of RNase contaminations. RNA oligonucleotides were as for EMSAs. 12.5 ng of plasmid (0.15 nM final concentration) were incubated in 20 µL of Invasion buffer (50 mM Tris-HCL pH 8.3, 10 mM DTT, 75 mM KCl, 0.01% BSA, 2% glycerol) containing 4 U of RNaseOUT (Thermo Fisher Scientific, Waltham) for 20 min on ice followed by 10 min at 25 °C, in the presence or absence of recombinant proteins. Labeled oligonucleotides were then added to the reactions at 1 nM concentration and incubated for 30 min at 25 °C. When specified, 60 U of RNaseH (Takara, Kusatu, Shiga, Japan) were also added in the reactions. For pulse-chase experiments, 1 nM of labeled oligonucleotides (hot) were incubated with 80 nM recombinant TRF2 for 20 min on ice followed by 10 min at 25 °C. Then, 12.5 ng of plasmid were added and reactions incubated for 30 min at 25 °C. Finally, 50 nM of un-labeled oligonucleotides (cold) were added to the reactions and the chase proceeded for 30, 60 or 120 min at 25 °C. All reactions were stopped by adding 1% SDS and 6 µg of Proteinase K and incubating for 15 min at 30 °C. Then 6× gel-loading buffer was added to a final concentration of 0.5×. Reaction products were fractionated by electrophoresis in 0.8% agarose gels, in 0.5× TBE. Gels were run at 45 V for 1 h at 25 °C and then dried and exposed to a phosphoimager screen. The radioactive signal detection and analysis were as for EMSAs. The number of independent replicates (n) is indicated in figure legends.
4.5. Statistical Analysis
For direct comparison of the two groups, we employed a paired or unpaired two-tailed Student’s t-test using GraphPad Prism (version 8.4.3, GraphPad software, San Diego, CA, USA). The p-values and used tests are indicated in figure legends.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




