
The Interplay between Aquaporin-1 and the Hypoxia-Inducible Factor 1α in a Lipopolysaccharide-Induced Lung Injury Model in Human Pulmonary Microvascular Endothelial Cells

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
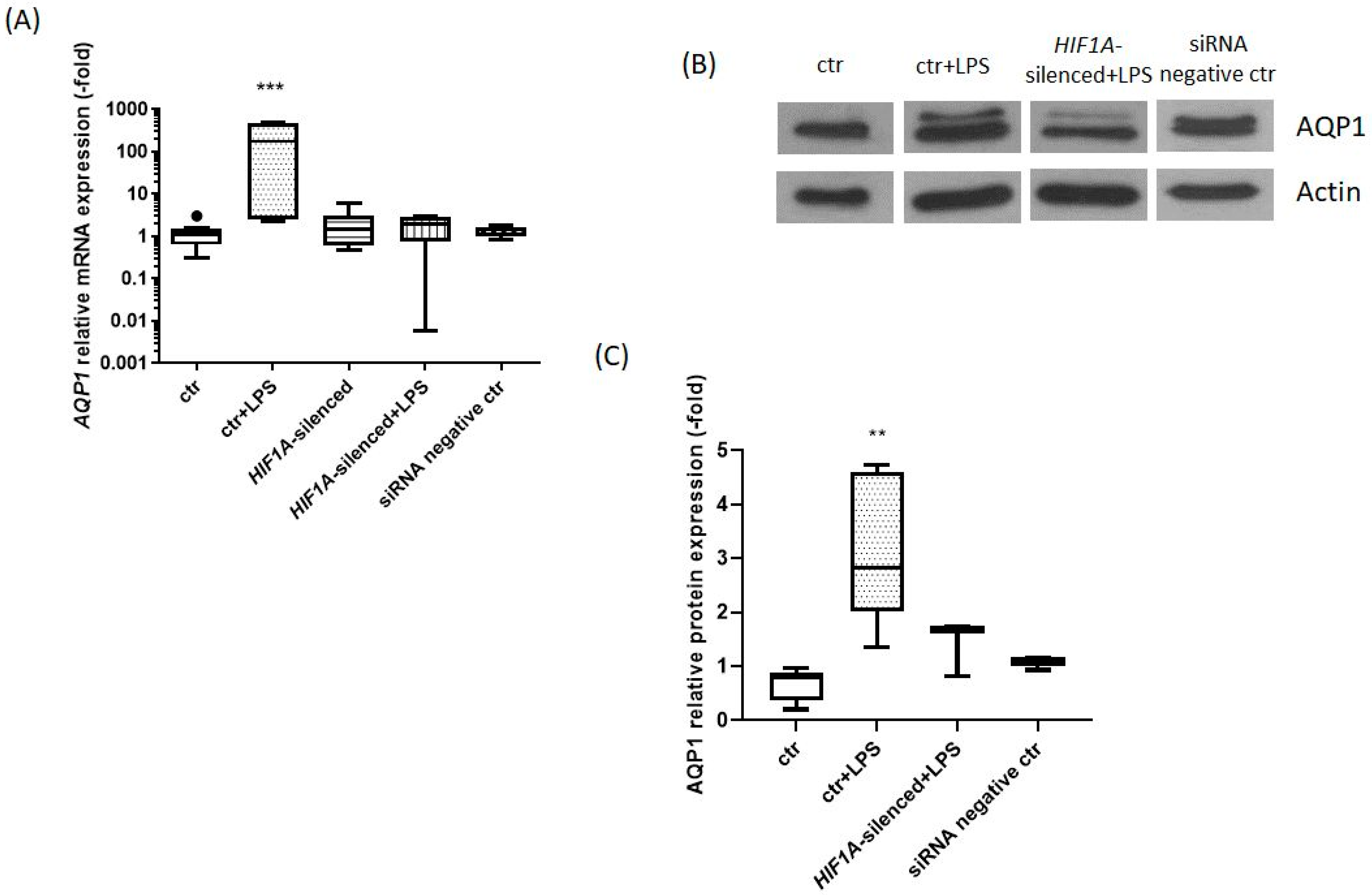
2.1. HIF1A Silencing Abrogates the LPS-Induced Increase in AQP1 mRNA and Protein Expression
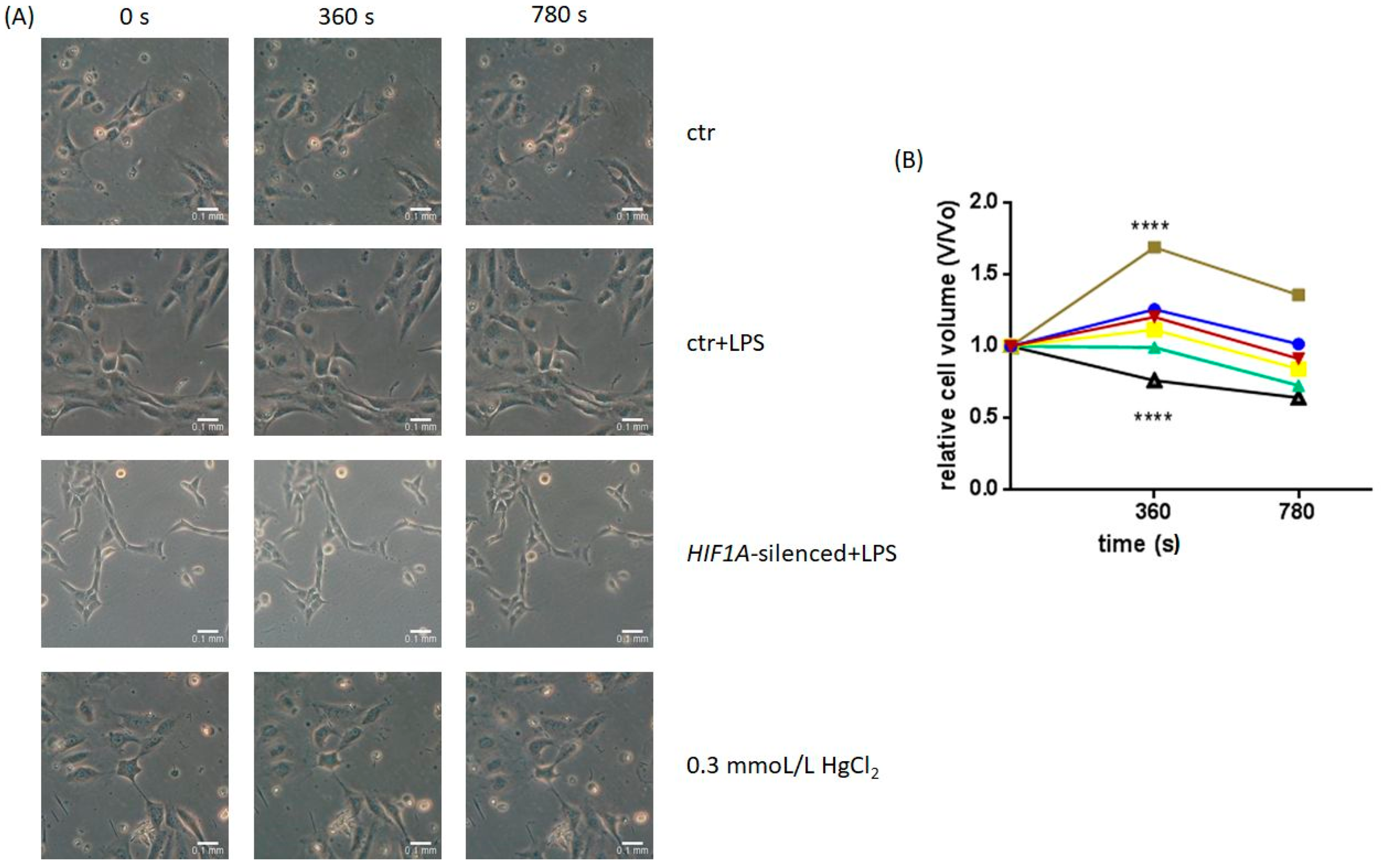
2.2. Osmotic Challenge of Untreated and HIF1A-Silenced HPMECs Exposed to LPS
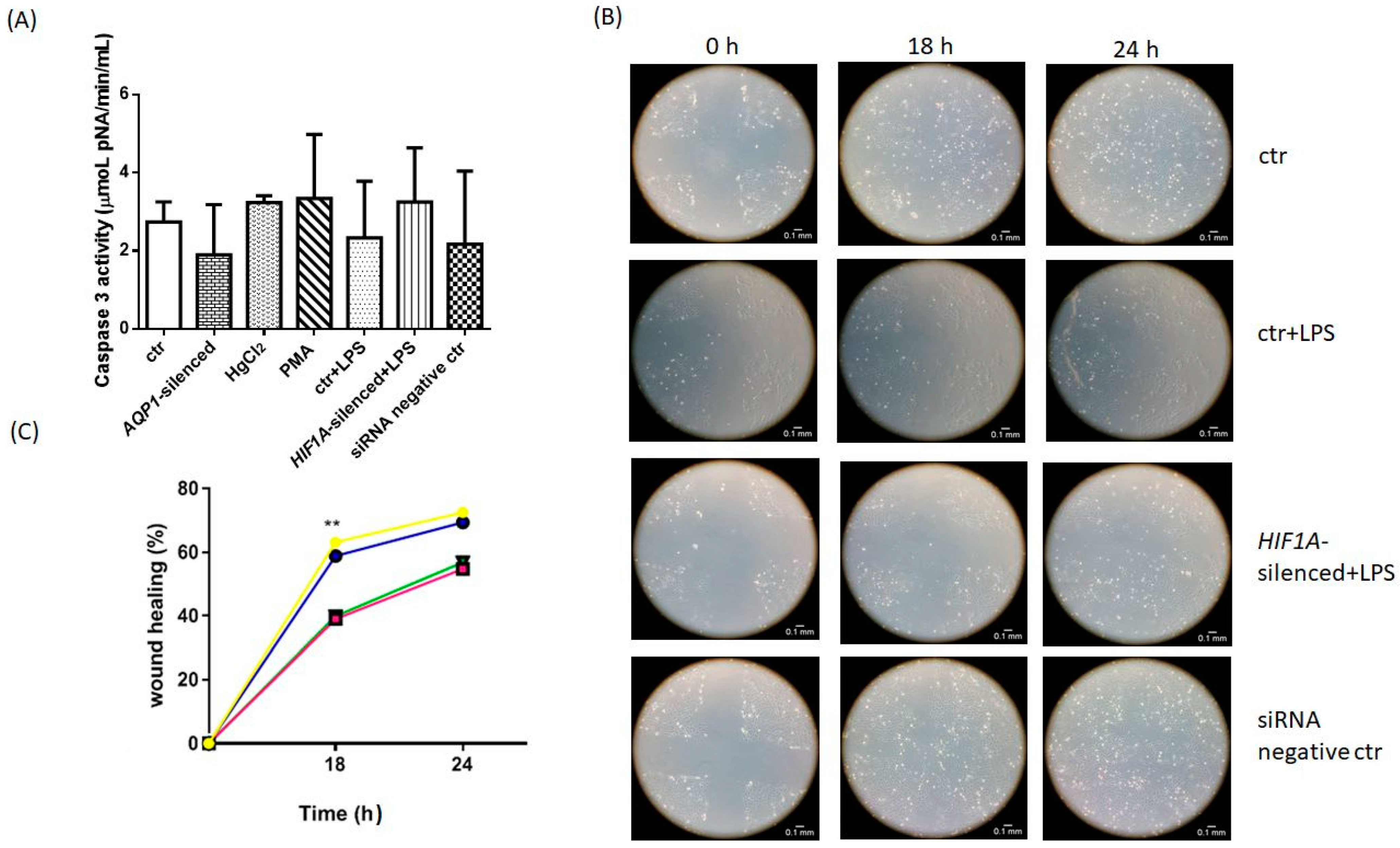
2.3. The Role of AQP1 in Apoptosis
2.4. Wound Healing Assay
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Force, T.A.D.T. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Keskinidou, C.; Vassiliou, A.G.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S.E. Mechanistic Understanding of Lung Inflammation: Recent Advances and Emerging Techniques. J. Inflamm. Res. 2022, 15, 3501–3546. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- Chalmers, S.; Khawaja, A.; Wieruszewski, P.M.; Gajic, O.; Odeyemi, Y. Diagnosis and treatment of acute pulmonary inflammation in critically ill patients: The role of inflammatory biomarkers. World J. Crit. Care Med. 2019, 8, 59–71. [Google Scholar] [CrossRef]
- Mobasheri, A.; Marples, D. Expression of the AQP-1 water channel in normal human tissues: A semiquantitative study using tissue microarray technology. Am. J. Physiol. Cell Physiol. 2004, 286, C529–C537. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Manitsopoulos, N.; Kardara, M.; Maniatis, N.A.; Orfanos, S.E.; Kotanidou, A. Differential Expression of Aquaporins in Experimental Models of Acute Lung Injury. In Vivo 2017, 31, 885–894. [Google Scholar] [CrossRef]
- Calero, C.; López-Campos, J.L.; Izquierdo, L.G.; Sánchez-Silva, R.; López-Villalobos, J.L.; Sáenz-Coronilla, F.J.; Arellano-Orden, E.; Montes-Worboys, A.; Echevarría, M. Expression of aquaporins in bronchial tissue and lung parenchyma of patients with chronic obstructive pulmonary disease. Multidiscip. Respir. Med. 2014, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Galán-Cobo, A.; Arellano-Orden, E.; Sánchez Silva, R.; López-Campos, J.L.; Gutiérrez Rivera, C.; Gómez Izquierdo, L.; Suárez-Luna, N.; Molina-Molina, M.; Rodríguez Portal, J.A.; Echevarría, M. The Expression of AQP1 IS Modified in Lung of Patients with Idiopathic Pulmonary Fibrosis: Addressing a Possible New Target. Front. Mol. Biosci. 2018, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Vassiliou, A.G.; Maniatis, N.A.; Orfanos, S.E.; Mastora, Z.; Jahaj, E.; Paparountas, T.; Armaganidis, A.; Roussos, C.; Aidinis, V.; Kotanidou, A. Induced expression and functional effects of aquaporin-1 in human leukocytes in sepsis. Crit. Care 2013, 17, R199. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Matthay, M.A.; Song, Y. Aquaporin water channels and lung physiology. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L867–L879. [Google Scholar] [CrossRef]
- Li, C.; Wang, W. Molecular Biology of Aquaporins. Adv. Exp. Med. Biol. 2017, 969, 1–34. [Google Scholar] [CrossRef]
- Verkman, A.S. Aquaporins in endothelia. Kidney Int. 2006, 69, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- Meli, R.; Pirozzi, C.; Pelagalli, A. New Perspectives on the Potential Role of Aquaporins (AQPs) in the Physiology of Inflammation. Front. Physiol. 2018, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef]
- Vanderhaeghen, T.; Vandewalle, J.; Libert, C. Hypoxia-inducible factors in metabolic reprogramming during sepsis. FEBS J. 2020, 287, 1478–1495. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Hasan, B.; Li, F.S.; Siyit, A.; Tuyghun, E.; Luo, J.H.; Upur, H.; Ablimit, A. Expression of aquaporins in the lungs of mice with acute injury caused by LPS treatment. Respir. Physiol. Neurobiol. 2014, 200, 40–45. [Google Scholar] [CrossRef]
- Su, X.; Song, Y.; Jiang, J.; Bai, C. The role of aquaporin-1 (AQP1) expression in a murine model of lipopolysaccharide-induced acute lung injury. Respir. Physiol. Neurobiol. 2004, 142, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.D.; Wu, X.Y.; Tao, B.D.; Wang, N.; Zhang, J. Protective effect and mechanism of hydrogen treatment on lung epithelial barrier dysfunction in rats with sepsis. Genet. Mol. Res. GMR 2016, 15, gmr.15016050. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, X.; Xia, X.; Zhang, Y. Estradiol attenuates LPS-induced acute lung injury via induction of aquaporins AQP1 and AQP5. Eur. J. Inflamm. 2021, 19, 20587392211049197. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE Signal Transduct. Knowl. Environ. 2005, 2005, re12. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E. Hypoxia-Inducible Factor Signaling in Inflammatory Lung Injury and Repair. Cells 2022, 11, 2. [Google Scholar] [CrossRef]
- McClendon, J.; Jansing, N.L.; Redente, E.F.; Gandjeva, A.; Ito, Y.; Colgan, S.P.; Ahmad, A.; Riches, D.W.H.; Chapman, H.A.; Mason, R.J.; et al. Hypoxia-Inducible Factor 1α Signaling Promotes Repair of the Alveolar Epithelium after Acute Lung Injury. Am. J. Pathol. 2017, 187, 1772–1786. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, X.; Zhao, D.X.; Yin, J.; Hu, G.; Evans, C.E.; Zhao, Y.Y. Endothelial Hypoxia-Inducible Factor-1α Is Required for Vascular Repair and Resolution of Inflammatory Lung Injury through Forkhead Box Protein M1. Am. J. Pathol. 2019, 189, 1664–1679. [Google Scholar] [CrossRef]
- Wu, G.; Xu, G.; Chen, D.-W.; Gao, W.-X.; Xiong, J.-Q.; Shen, H.-Y.; Gao, Y.-Q. Hypoxia Exacerbates Inflammatory Acute Lung Injury via the Toll-Like Receptor 4 Signaling Pathway. Front. Immunol. 2018, 9, 1667. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Cejudo-Martin, P.; Doedens, A.; Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J. Immunol. 2007, 178, 7516–7519. [Google Scholar] [CrossRef]
- Yeh, C.H.; Cho, W.; So, E.C.; Chu, C.C.; Lin, M.C.; Wang, J.J.; Hsing, C.H. Propofol inhibits lipopolysaccharide-induced lung epithelial cell injury by reducing hypoxia-inducible factor-1alpha expression. Br. J. Anaesth. 2011, 106, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Cao, F.; Liu, L.; Zhang, B.; Wang, Y.; Dong, H.; Cui, Y.; Dong, M.; Xu, D.; Liu, Y.; et al. Tanshinone IIA-induced attenuation of lung injury in endotoxemic mice is associated with reduction of hypoxia-inducible factor 1α expression. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shan, C.; Wu, Z.; Yu, H.; Yang, A.; Tan, B. Correction to: Emodin alleviated pulmonary inflammation in rats with LPS-induced acute lung injury through inhibiting the mTOR/HIF-1α/VEGF signaling pathway. Inflamm. Res. 2020, 69, 711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiong, Y.; Lu, L.X.; Wang, H.; Zhang, Y.F.; Fang, F.; Song, Y.L.; Jiang, H. AQP1 expression alterations affect morphology and water transport in Schwann cells and hypoxia-induced up-regulation of AQP1 occurs in a HIF-1alpha-dependent manner. Neuroscience 2013, 252, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Yu, G.; Liu, Q.; Jin, Y. Cytoprotective effect of aquaporin 1 against lipopolysaccharide-induced apoptosis and inflammation of renal epithelial HK-2 cells. Exp. Ther. Med. 2018, 15, 4243–4252. [Google Scholar] [CrossRef]
- Jablonski, E.M.; Webb, A.N.; McConnell, N.A.; Riley, M.C.; Hughes, F.M., Jr. Plasma membrane aquaporin activity can affect the rate of apoptosis but is inhibited after apoptotic volume decrease. Am. J. Physiol. Cell Physiol. 2004, 286, C975–C985. [Google Scholar] [CrossRef]
- Schuoler, C.; Haider, T.J.; Leuenberger, C.; Vogel, J.; Ostergaard, L.; Kwapiszewska, G.; Kohler, M.; Gassmann, M.; Huber, L.C.; Brock, M. Aquaporin 1 controls the functional phenotype of pulmonary smooth muscle cells in hypoxia-induced pulmonary hypertension. Basic Res. Cardiol. 2017, 112, 30. [Google Scholar] [CrossRef]
- Michaelis, U.R. Mechanisms of endothelial cell migration. Cell. Mol. Life Sci. 2014, 71, 4131–4148. [Google Scholar] [CrossRef]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef]
- Dorward, H.S.; Du, A.; Bruhn, M.A.; Wrin, J.; Pei, J.V.; Evdokiou, A.; Price, T.J.; Yool, A.J.; Hardingham, J.E. Pharmacological blockade of aquaporin-1 water channel by AqB013 restricts migration and invasiveness of colon cancer cells and prevents endothelial tube formation in vitro. J. Exp. Clin. Cancer Res. 2016, 35, 36. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Saadoun, S.; Verkman, A.S. Aquaporins and cell migration. Pflug. Arch. Eur. J. Physiol. 2008, 456, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y. Aquaporin-1 activity of plasma membrane affects HT20 colon cancer cell migration. IUBMB Life 2009, 61, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Dong, J. Aquaporin 1 promotes the proliferation and migration of lung cancer cell in vitro. Oncol. Rep. 2015, 34, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Unger, L.; Salman, M.M.; Kitchen, P.; Bill, R.M.; Yool, A.J. Signaling Mechanisms and Pharmacological Modulators Governing Diverse Aquaporin Functions in Human Health and Disease. Int. J. Mol. Sci. 2022, 23, 3. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, W.; Hu, X. Different concentrations of lipopolysaccharide regulate barrier function through the PI3K/Akt signalling pathway in human pulmonary microvascular endothelial cells. Sci. Rep. 2018, 8, 9963. [Google Scholar] [CrossRef]
- Krump-Konvalinkova, V.; Bittinger, F.; Unger, R.E.; Peters, K.; Lehr, H.-A.; Kirkpatrick, C.J. Generation of Human Pulmonary Microvascular Endothelial Cell Lines. Lab. Investig. 2001, 81, 1717–1727. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lleu, P.L.; Rebel, G. Interference of Good’s buffers and other biological buffers with protein determination. Anal. Biochem. 1991, 192, 215–218. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample | Caspase 3 Activity (μmoL pNA/min/mL) |
|---|---|
| ctr | 2.74 ± 1.4 |
| AQP1-silenced | 1.90 ± 1.3 |
| HgCl2 | 3.24 ± 0.18 |
| PMA | 3.34 ± 1.6 |
| ctr + LPS | 2.33 ± 1.5 |
| HIF1A-silenced + LPS | 2.77 ± 1.6 |
| siRNA negative ctr | 2.18 ± 1.9 |
| Sample | 18 h (%) | 24 h (%) |
|---|---|---|
| ctr | 63 ± 20 | 72 ± 19 |
| ctr + LPS | 39 ± 17 ** | 55 ± 20 |
| HIF1A-silenced + LPS | 40 ± 16 ** | 57 ± 19 |
| siRNA negative ctr | 59 ± 22 | 69 ± 20 |
| Gene | Forward Primer | Reverse Primer | Amplicon Size (Base Pairs) |
|---|---|---|---|
| AQP1 | 5′-CTG-GGC-ATC-GAG-ATC-ATC-GG-3′ | 5′-ATC-CCA-CAG-CCA-GTG-TAG-TCA-3′ | 158 |
| HIF1A | 5′-GGC-GCG-AAC-GAC-AAG-AAA-AAG-3′ | 5′-CCT-TAT-CAA-GAT-GCG-AAC-TCA-CA-3′ | 154 |
| GAPDH | 5′-ATG-GGG-AAG-GTG-AAG-GTC-G-3′ | 5′-TAC-ATG-AGG-GCA-CGG-AAG-ATG-3′ | 108 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keskinidou, C.; Lotsios, N.S.; Vassiliou, A.G.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S.E. The Interplay between Aquaporin-1 and the Hypoxia-Inducible Factor 1α in a Lipopolysaccharide-Induced Lung Injury Model in Human Pulmonary Microvascular Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 10588. https://doi.org/10.3390/ijms231810588
Keskinidou C, Lotsios NS, Vassiliou AG, Dimopoulou I, Kotanidou A, Orfanos SE. The Interplay between Aquaporin-1 and the Hypoxia-Inducible Factor 1α in a Lipopolysaccharide-Induced Lung Injury Model in Human Pulmonary Microvascular Endothelial Cells. International Journal of Molecular Sciences. 2022; 23(18):10588. https://doi.org/10.3390/ijms231810588
Chicago/Turabian StyleKeskinidou, Chrysi, Nikolaos S. Lotsios, Alice G. Vassiliou, Ioanna Dimopoulou, Anastasia Kotanidou, and Stylianos E. Orfanos. 2022. "The Interplay between Aquaporin-1 and the Hypoxia-Inducible Factor 1α in a Lipopolysaccharide-Induced Lung Injury Model in Human Pulmonary Microvascular Endothelial Cells" International Journal of Molecular Sciences 23, no. 18: 10588. https://doi.org/10.3390/ijms231810588
APA StyleKeskinidou, C., Lotsios, N. S., Vassiliou, A. G., Dimopoulou, I., Kotanidou, A., & Orfanos, S. E. (2022). The Interplay between Aquaporin-1 and the Hypoxia-Inducible Factor 1α in a Lipopolysaccharide-Induced Lung Injury Model in Human Pulmonary Microvascular Endothelial Cells. International Journal of Molecular Sciences, 23(18), 10588. https://doi.org/10.3390/ijms231810588