

Upregulated Apelin Signaling in Pancreatic Cancer Activates Oncogenic Signaling Pathways to Promote Tumor Development

 , , add
Show full author list
, , add
Show full author list
Abstract
:1. Introduction
2. Results
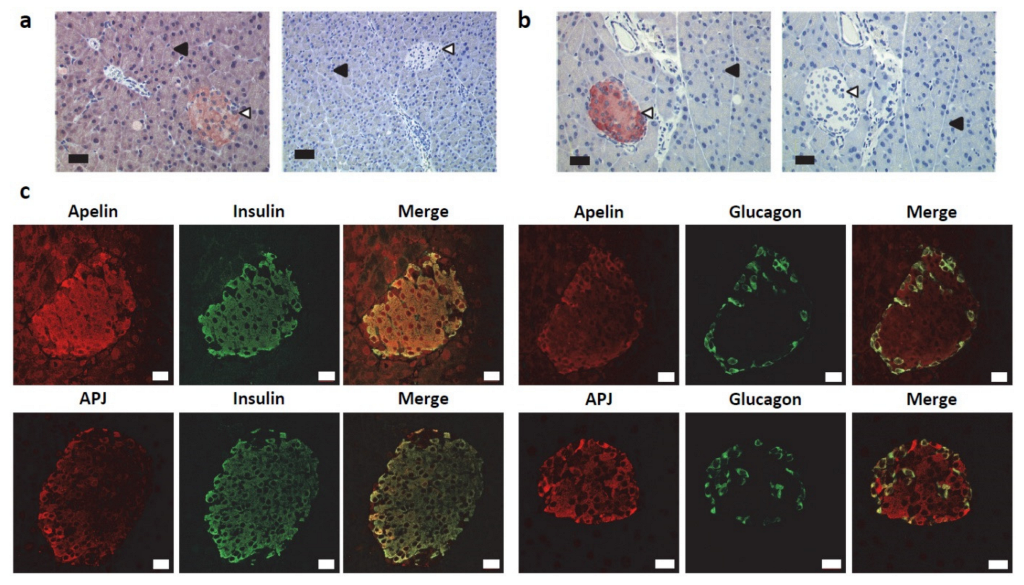
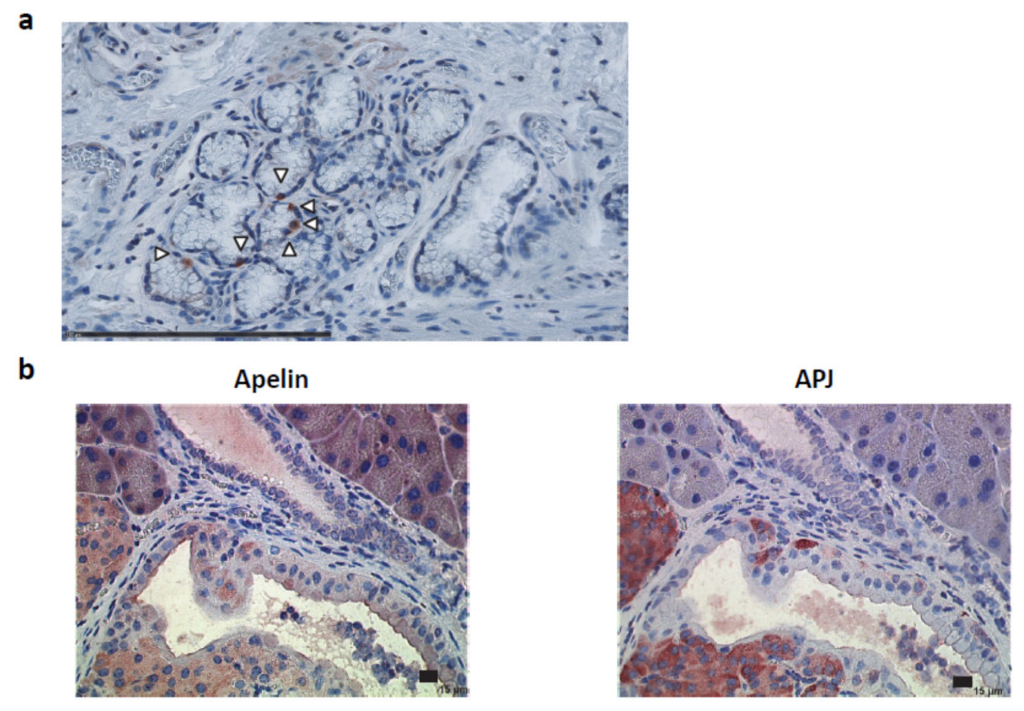
2.1. Apelin and APJ Are Differentially Expressed in Normal Pancreas
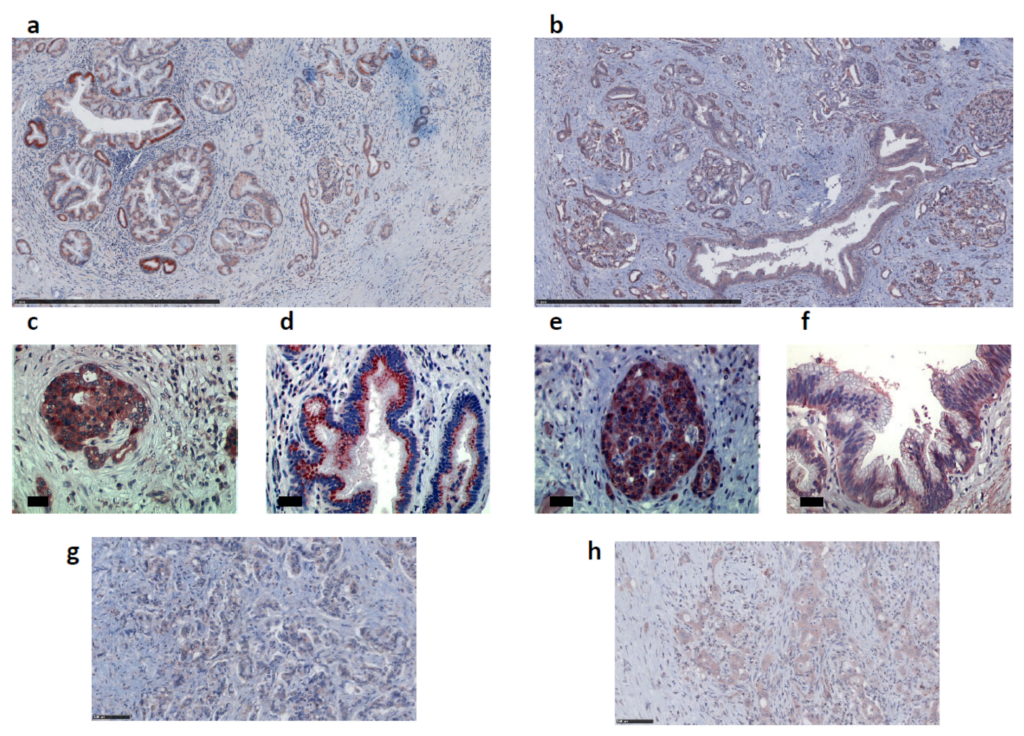
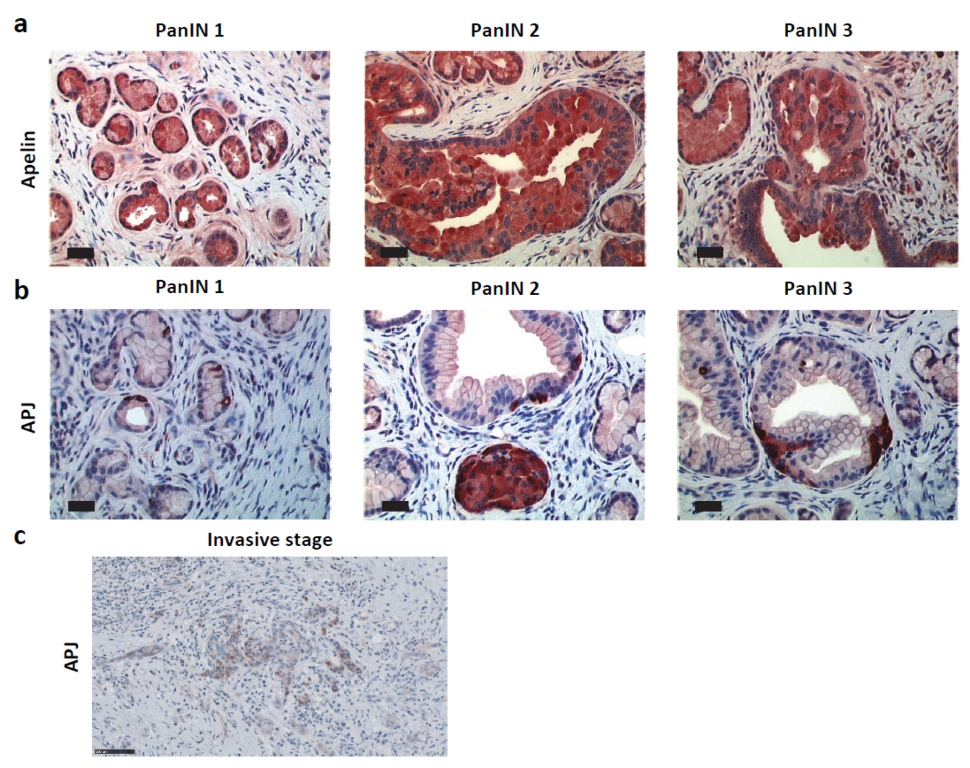
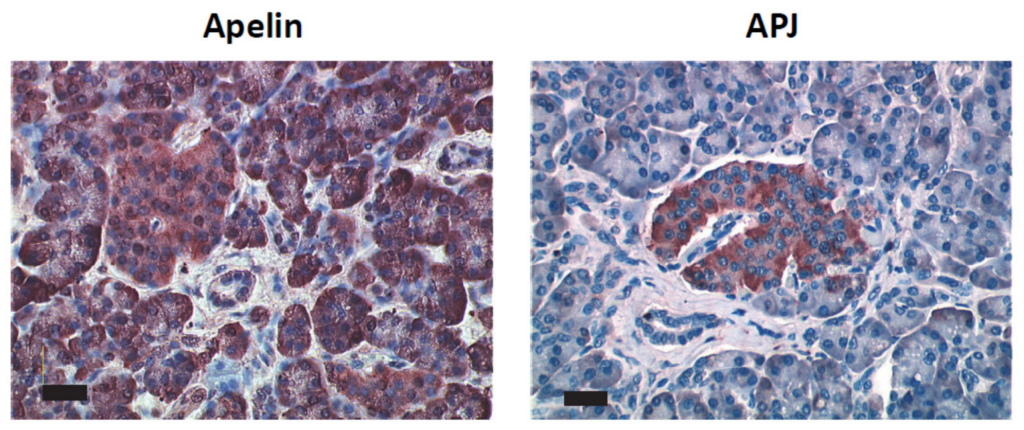
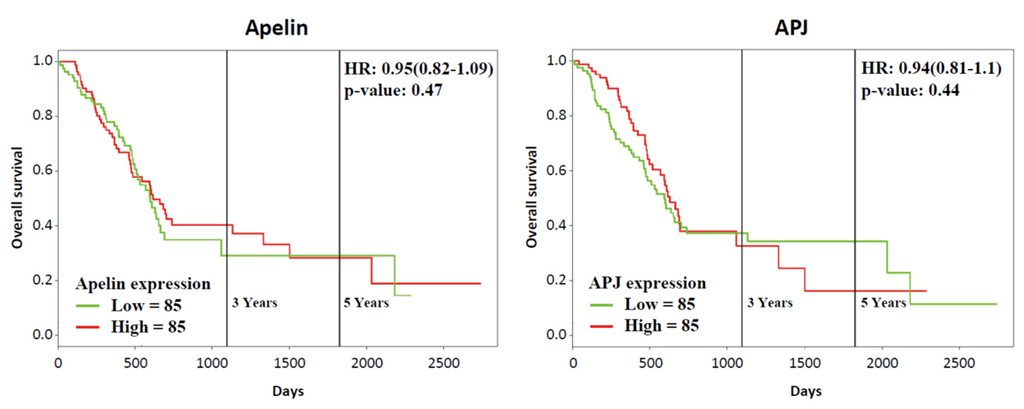
2.2. Apelin and APJ Are Expressed in Human and Murine Premalignant and Malignant Pancreatic Lesions
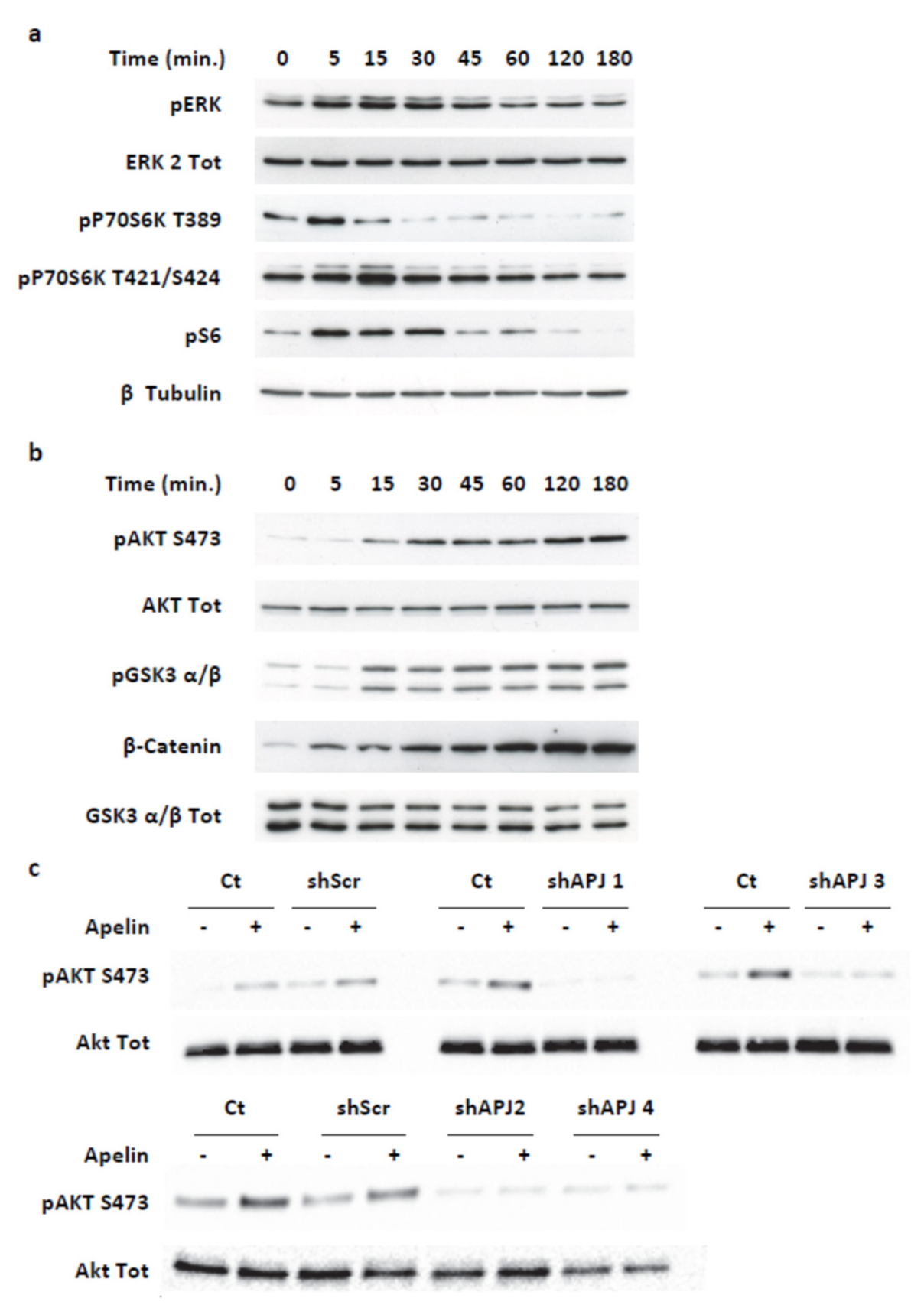
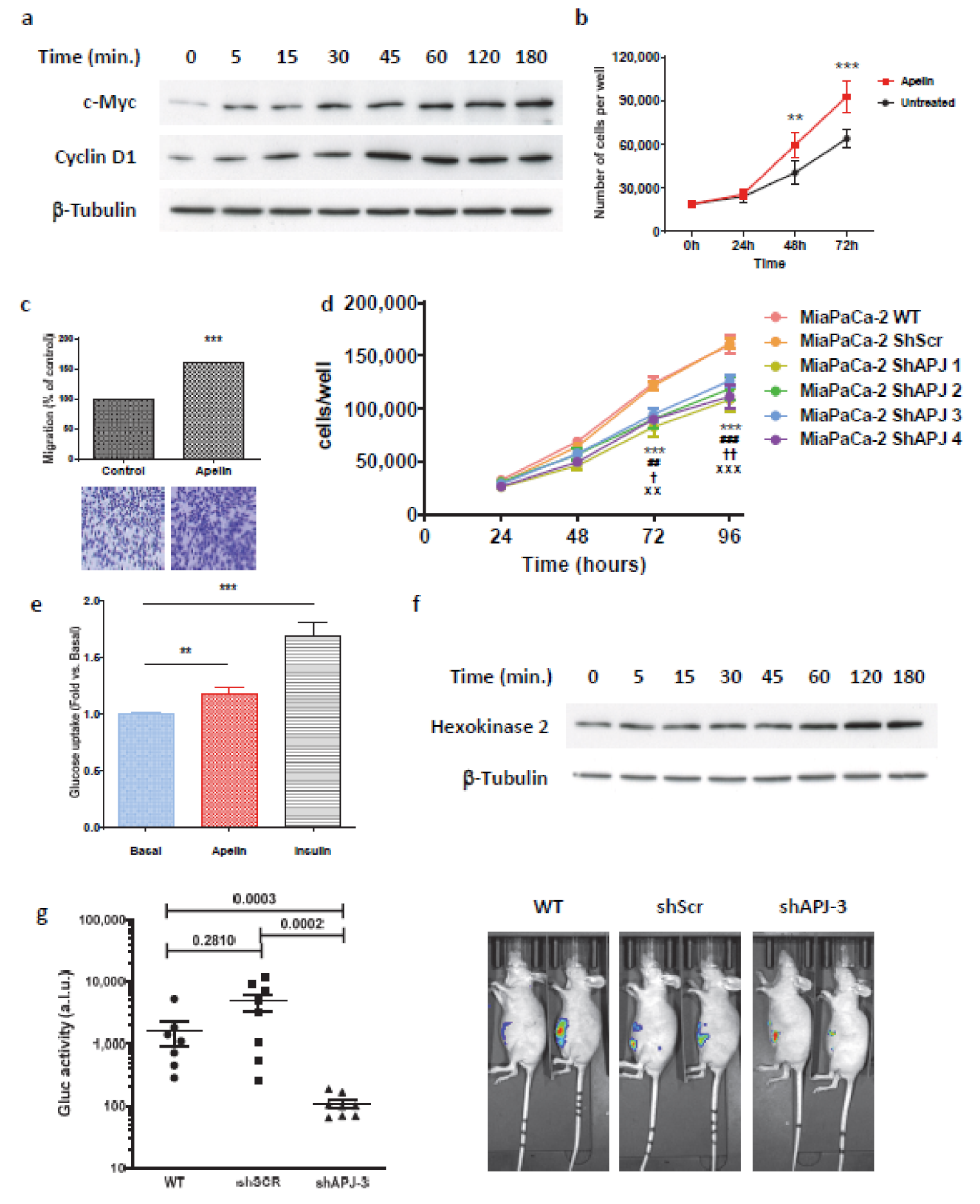
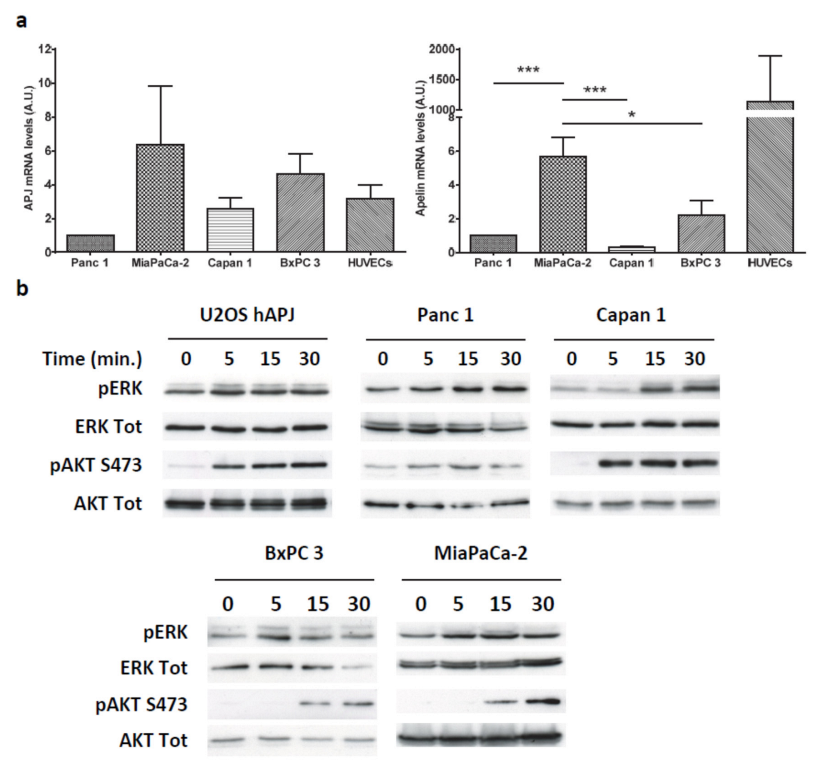
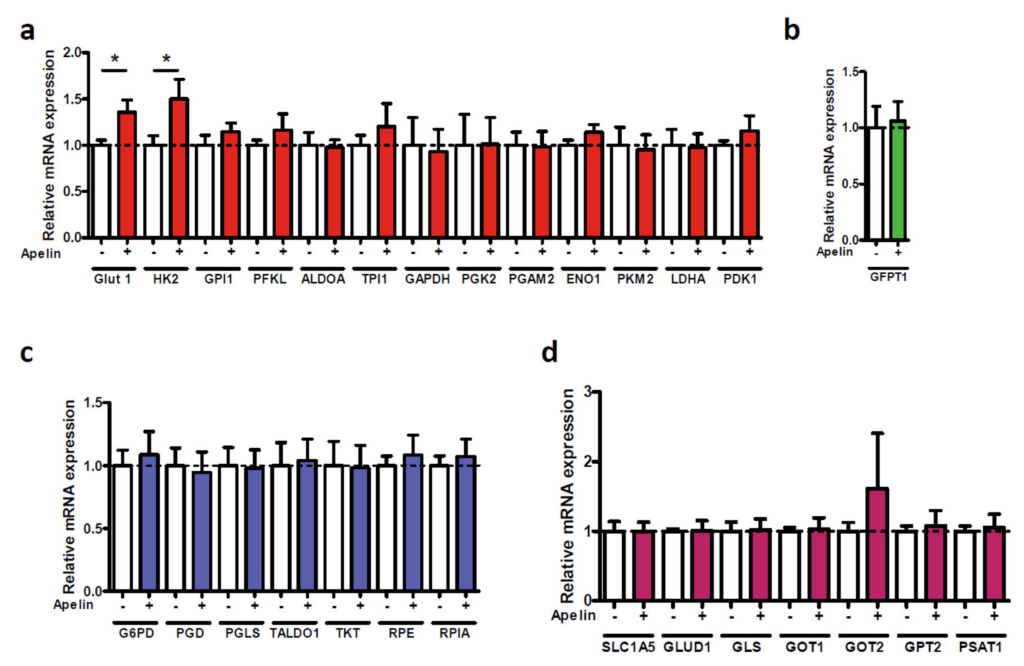
2.3. Apelin Induces Different Signaling Pathways in Human Pancreatic Tumor Cells
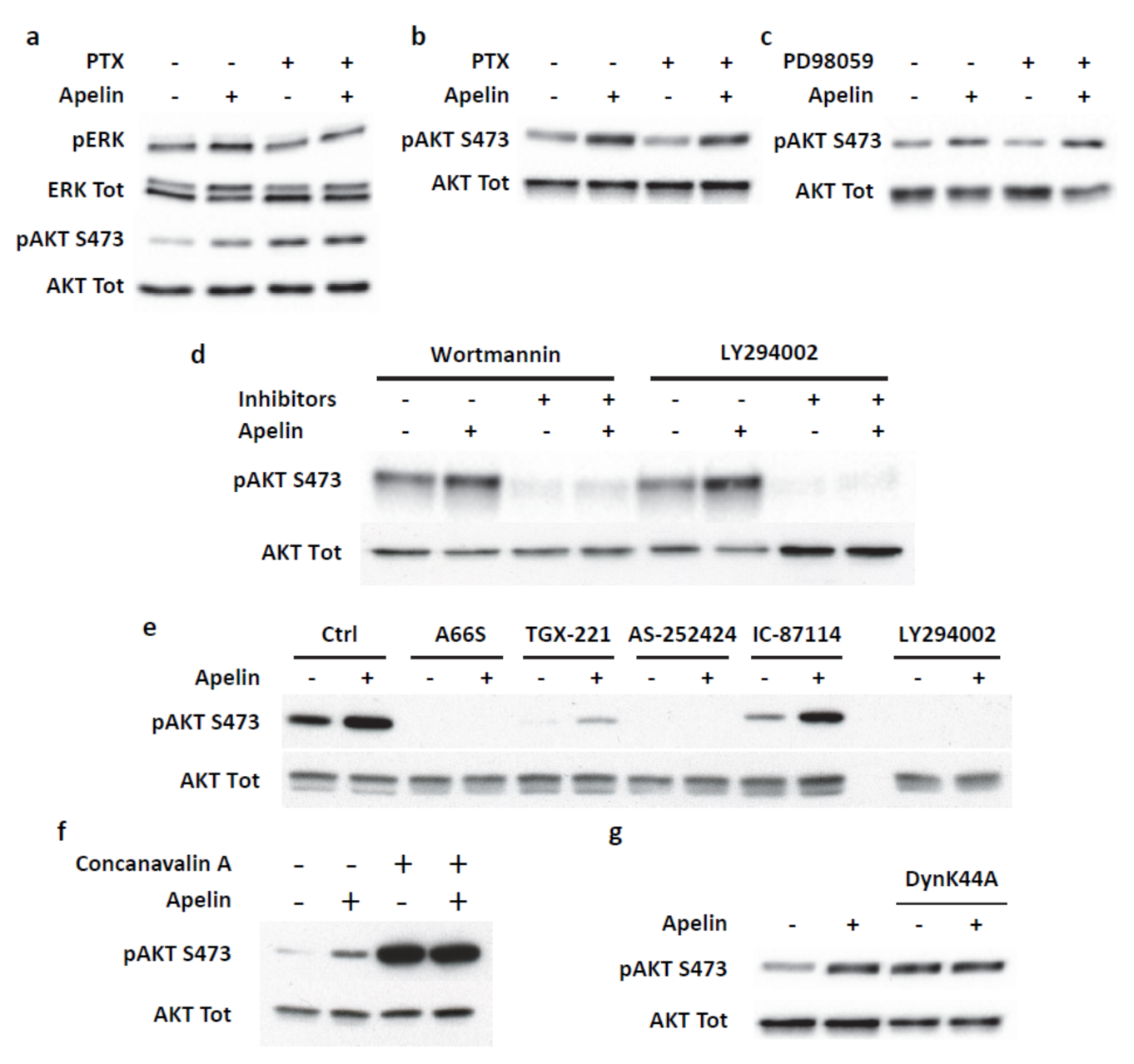
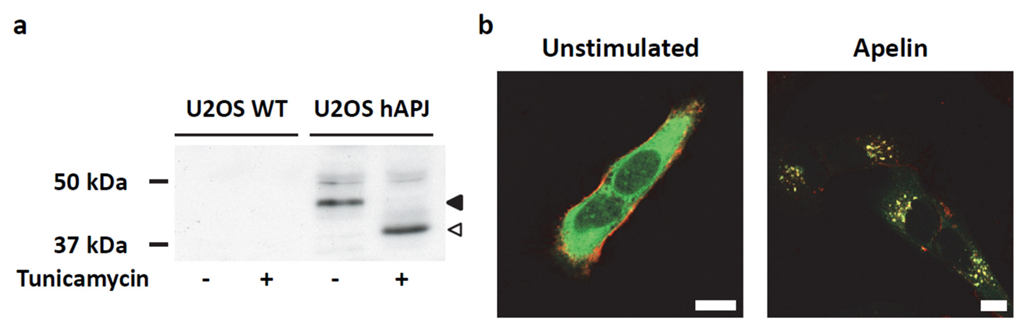
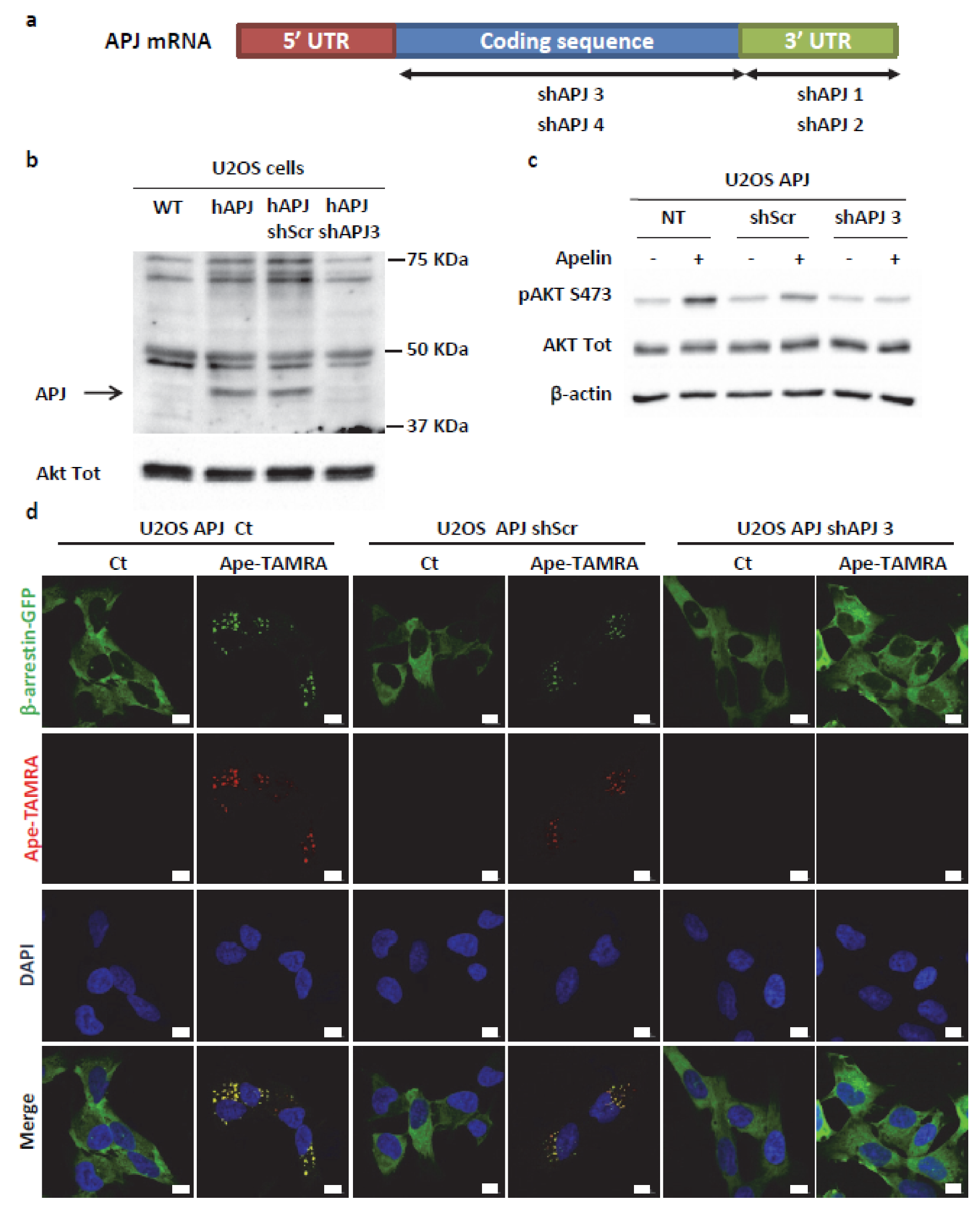
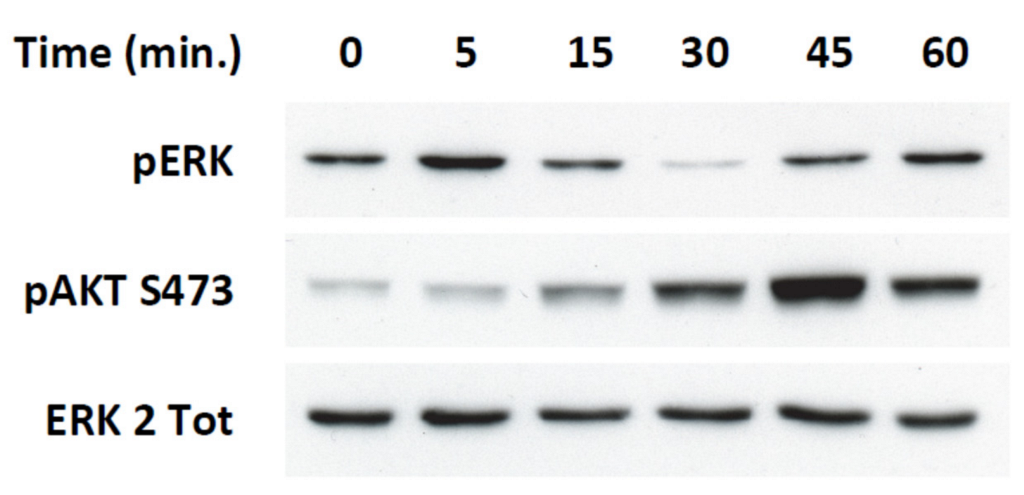
2.4. Transient ERKs Phosphorylation Induced by Apelin Depends on G-Protein Pathway Whereas Long Lasting Akt Stimulation Relies on APJ Internalization
2.5. Secreted Apelin by Tumor Cells Induced Pancreatic Cancer Burden
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Mice
4.3. Cell Culture
4.4. Proliferation and Migration Assay
4.5. Immunohistochemistry
4.6. Western Blot Analysis
4.7. Tumor Model
4.8. Fluorescence Microscopy
4.9. Glucose Uptake
4.10. Quantitative Real-Time PCR
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A








References
- Bouvier, A.M.; Bossard, N.; Colonna, M.; Garcia-Velasco, A.; Carulla, M.; Manfredi, S. Trends in net survival from pancreatic cancer in six European Latin countries: Results from the SUDCAN population-based study. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. (ECP) 2017, 26, S63–S69. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J. Natl. Cancer Inst. 2015, 107, dju413. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.; Tsui, L.C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Masri, B.; Knibiehler, B.; Audigier, Y. Apelin signalling: A promising pathway from cloning to pharmacology. Cell Signal. 2005, 17, 415–426. [Google Scholar] [CrossRef]
- Masri, B.; Lahlou, H.; Mazarguil, H.; Knibiehler, B.; Audigier, Y. Apelin (65–77) activates extracellular signal-regulated kinases via a PTX-sensitive G protein. Biochem. Biophys. Res. Commun. 2002, 290, 539–545. [Google Scholar] [CrossRef]
- Masri, B.; Morin, N.; Pedebernade, L.; Knibiehler, B.; Audigier, Y. The apelin receptor is coupled to Gi1 or Gi2 protein and is differentially desensitized by apelin fragments. J. Biol. Chem. 2006, 281, 18317–18326. [Google Scholar] [CrossRef]
- Chaves-Almagro, C.; Castan-Laurell, I.; Dray, C.; Knauf, C.; Valet, P.; Masri, B. Apelin receptors: From signaling to antidiabetic strategy. Eur. J. Pharm. 2015, 763 Pt B, 149–159. [Google Scholar] [CrossRef]
- Masri, B.; Morin, N.; Cornu, M.; Knibiehler, B.; Audigier, Y. Apelin (65-77) activates p70 S6 kinase and is mitogenic for umbilical endothelial cells. FASEB J. 2004, 18, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Szokodi, I.; Tavi, P.; Foldes, G.; Voutilainen-Myllyla, S.; Ilves, M.; Tokola, H.; Pikkarainen, S.; Piuhola, J.; Rysa, J.; Toth, M.; et al. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ. Res. 2002, 91, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Kasai, A.; Shintani, N.; Oda, M.; Kakuda, M.; Hashimoto, H.; Matsuda, T.; Hinuma, S.; Baba, A. Apelin is a novel angiogenic factor in retinal endothelial cells. Biochem. Biophys. Res. Commun. 2004, 325, 395–400. [Google Scholar] [CrossRef]
- Cox, C.M.; D’Agostino, S.L.; Miller, M.K.; Heimark, R.L.; Krieg, P.A. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev. Biol. 2006, 296, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Dray, C.; Knauf, C.; Daviaud, D.; Waget, A.; Boucher, J.; Buleon, M.; Cani, P.D.; Attane, C.; Guigne, C.; Carpene, C.; et al. Apelin stimulates glucose utilization in normal and obese insulin-resistant mice. Cell Metab. 2008, 8, 437–445. [Google Scholar] [CrossRef]
- Ringstrom, C.; Nitert, M.D.; Bennet, H.; Fex, M.; Valet, P.; Rehfeld, J.F.; Friis-Hansen, L.; Wierup, N. Apelin is a novel islet peptide. Regul. Pept. 2010, 162, 44–51. [Google Scholar] [CrossRef]
- Sorhede Winzell, M.; Magnusson, C.; Ahren, B. The apj receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul. Pept. 2005, 131, 12–17. [Google Scholar] [CrossRef]
- Han, S.; Englander, E.W.; Gomez, G.A.; Rastellini, C.; Quertermous, T.; Kundu, R.K.; Greeley, G.H., Jr. Pancreatic Islet APJ Deletion Reduces Islet Density and Glucose Tolerance in Mice. Endocrinology 2015, 156, 2451–2460. [Google Scholar] [CrossRef]
- Han, S.; Englander, E.W.; Gomez, G.A.; Aronson, J.F.; Rastellini, C.; Garofalo, R.P.; Kolli, D.; Quertermous, T.; Kundu, R.; Greeley, G.H., Jr. Pancreatitis activates pancreatic apelin-APJ axis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G139–G150. [Google Scholar] [CrossRef]
- Han, S.; Englander, E.W.; Gomez, G.A.; Greeley, G.H., Jr. Apelin Regulates Nuclear Factor-kappaB’s Involvement in the Inflammatory Response of Pancreatitis. Pancreas 2017, 46, 64–70. [Google Scholar] [CrossRef]
- Sorli, S.C.; Le Gonidec, S.; Knibiehler, B.; Audigier, Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene 2007, 26, 7692–7699. [Google Scholar] [CrossRef] [PubMed]
- Berta, J.; Kenessey, I.; Dobos, J.; Tovari, J.; Klepetko, W.; Jan Ankersmit, H.; Hegedus, B.; Renyi-Vamos, F.; Varga, J.; Lorincz, Z.; et al. Apelin expression in human non-small cell lung cancer: Role in angiogenesis and prognosis. J. Thorac. Oncol. 2010, 5, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.; Kim, Y.H.; Sung, H.J.; Li, H.Y.; Yoo, C.W.; Kim, J.Y.; Park, J.Y.; Lee, U.L.; Nam, B.H.; Kim, E.O.; et al. Hypoxia-induced up-regulation of apelin is associated with a poor prognosis in oral squamous cell carcinoma patients. Oral Oncol. 2012, 48, 500–506. [Google Scholar] [CrossRef]
- Kalin, R.E.; Kretz, M.P.; Meyer, A.M.; Kispert, A.; Heppner, F.L.; Brandli, A.W. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumor angiogenesis. Dev. Biol. 2007, 305, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Picault, F.X.; Chaves-Almagro, C.; Projetti, F.; Prats, H.; Masri, B.; Audigier, Y. Tumour co-expression of apelin and its receptor is the basis of an autocrine loop involved in the growth of colon adenocarcinomas. Eur. J. Cancer 2014, 50, 663–674. [Google Scholar] [CrossRef]
- Lacquaniti, A.; Altavilla, G.; Picone, A.; Donato, V.; Chirico, V.; Mondello, P.; Aloisi, C.; Marabello, G.; Loddo, S.; Buemi, A.; et al. Apelin beyond kidney failure and hyponatremia: A useful biomarker for cancer disease progression evaluation. Clin. Exp. Med. 2014, 15, 97–105. [Google Scholar] [CrossRef]
- Berta, J.; Hoda, M.A.; Laszlo, V.; Rozsas, A.; Garay, T.; Torok, S.; Grusch, M.; Berger, W.; Paku, S.; Renyi-Vamos, F.; et al. Apelin promotes lymphangiogenesis and lymph node metastasis. Oncotarget 2014, 5, 4426–4437. [Google Scholar] [CrossRef]
- Sorli, S.C.; van den Berghe, L.; Masri, B.; Knibiehler, B.; Audigier, Y. Therapeutic potential of interfering with apelin signalling. Drug Discov. Today 2006, 11, 1100–1106. [Google Scholar] [CrossRef]
- Puffer, B.A.; Sharron, M.; Coughlan, C.M.; Baribaud, F.; McManus, C.M.; Lee, B.; David, J.; Price, K.; Horuk, R.; Tsang, M.; et al. Expression and coreceptor function of APJ for primate immunodeficiency viruses. Virology 2000, 276, 435–444. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Le Gonidec, S.; Chaves-Almagro, C.; Bai, Y.; Kang, H.J.; Smith, A.; Wanecq, E.; Huang, X.P.; Prats, H.; Knibiehler, B.; Roth, B.L.; et al. Protamine is an antagonist of apelin receptor, and its activity is reversed by heparin. FASEB J. 2017, 31, 2507–2519. [Google Scholar] [CrossRef] [PubMed]
- Chaussade, C.; Rewcastle, G.W.; Kendall, J.D.; Denny, W.A.; Cho, K.; Gronning, L.M.; Chong, M.L.; Anagnostou, S.H.; Jackson, S.P.; Daniele, N.; et al. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem. J. 2007, 404, 449–458. [Google Scholar] [CrossRef]
- Takaki, R.; Ono, J.; Nakamura, M.; Yokogawa, Y.; Kumae, S.; Hiraoka, T.; Yamaguchi, K.; Hamaguchi, K.; Uchida, S. Isolation of glucagon-secreting cell lines by cloning insulinoma cells. Vitr. Cell. Dev. Biol. J. Tissue Cult. Assoc. 1986, 22 Pt 1, 120–126. [Google Scholar] [CrossRef]
- Barreto, S.G.; Michael, M.Z.; Keating, D.J. Islets and pancreatic ductal adenocarcinoma-An opportunity for translational research from the ‘Bench to the Bedside’. Pancreatology 2020, 20, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Larsson, J.; Adrian, T.E.; Gasslander, T.; Permert, J. In vitro influences between pancreatic adenocarcinoma cells and pancreatic islets. J. Surg. Res. 1998, 79, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Masri, B.; Daviaud, D.; Gesta, S.; Guigne, C.; Mazzucotelli, A.; Castan-Laurell, I.; Tack, I.; Knibiehler, B.; Carpene, C.; et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology 2005, 146, 1764–1771. [Google Scholar] [CrossRef]
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schonhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes Dev. 2014, 28, 2621–2635. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Edling, C.E.; Selvaggi, F.; Buus, R.; Maffucci, T.; Di Sebastiano, P.; Friess, H.; Innocenti, P.; Kocher, H.M.; Falasca, M. Key role of phosphoinositide 3-kinase class IB in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 4928–4937. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Mancinelli, G.; Cordoba-Chacon, J.; Viswakarma, N.; Castellanos, K.; Grimaldo, S.; Kumar, S.; Principe, D.; Dorman, M.J.; McKinney, R.; et al. p110gamma deficiency protects against pancreatic carcinogenesis yet predisposes to diet-induced hepatotoxicity. Proc. Natl. Acad. Sci. USA 2019, 116, 14724–14733. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; DeFea, K.A. Protease-activated receptor-2 simultaneously directs beta-arrestin-1-dependent inhibition and Galphaq-dependent activation of phosphatidylinositol 3-kinase. Biochemistry 2006, 45, 9374–9385. [Google Scholar] [CrossRef]
- Wang, P.; Kumar, P.; Wang, C.; Defea, K.A. Differential regulation of class IA phosphoinositide 3-kinase catalytic subunits p110 alpha and beta by protease-activated receptor 2 and beta-arrestins. Biochem. J. 2007, 408, 221–230. [Google Scholar] [CrossRef]
- Arakaki, A.K.S.; Pan, W.A.; Trejo, J. GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling. Int. J. Mol. Sci. 2018, 19, 1886. [Google Scholar] [CrossRef]
- English, E.J.; Mahn, S.A.; Marchese, A. Endocytosis is required for CXC chemokine receptor type 4 (CXCR4)-mediated Akt activation and antiapoptotic signaling. J. Biol. Chem. 2018, 293, 11470–11480. [Google Scholar] [CrossRef]
- Pampillo, M.; Camuso, N.; Taylor, J.E.; Szereszewski, J.M.; Ahow, M.R.; Zajac, M.; Millar, R.P.; Bhattacharya, M.; Babwah, A.V. Regulation of GPR54 signaling by GRK2 and {beta}-arrestin. Mol. Endocrinol. 2009, 23, 2060–2074. [Google Scholar] [CrossRef]
- Fu, Q.; Chen, Z.; Gong, X.; Cai, Y.; Chen, Y.; Ma, X.; Zhu, R.; Jin, J. beta-Catenin expression is regulated by an IRES-dependent mechanism and stimulated by paclitaxel in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 21–27. [Google Scholar] [CrossRef]
- Holmes, B.; Lee, J.; Landon, K.A.; Benavides-Serrato, A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Mechanistic Target of Rapamycin (mTOR) Inhibition Synergizes with Reduced Internal Ribosome Entry Site (IRES)-mediated Translation of Cyclin D1 and c-MYC mRNAs to Treat Glioblastoma. J. Biol. Chem. 2016, 291, 14146–14159. [Google Scholar] [CrossRef] [Green Version]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of beta-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Tamaki, N.; Honda, T.; Torizuka, T.; Kimura, T.; Inokuma, T.; Ohshio, G.; Hosotani, R.; Imamura, M.; Konishi, J. Expression of glucose transporters in human pancreatic tumors compared with increased FDG accumulation in PET study. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1997, 38, 1337–1344. [Google Scholar]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2017, 8, 56081–56094. [Google Scholar] [CrossRef]
- Ogawa, H.; Nagano, H.; Konno, M.; Eguchi, H.; Koseki, J.; Kawamoto, K.; Nishida, N.; Colvin, H.; Tomokuni, A.; Tomimaru, Y.; et al. The combination of the expression of hexokinase 2 and pyruvate kinase M2 is a prognostic marker in patients with pancreatic cancer. Mol. Clin. Oncol. 2015, 3, 563–571. [Google Scholar] [CrossRef]
- Sicard, F.; Gayral, M.; Lulka, H.; Buscail, L.; Cordelier, P. Targeting miR-21 for the therapy of pancreatic cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 986–994. [Google Scholar] [CrossRef]
- Jiang, S.H.; Li, J.; Dong, F.Y.; Yang, J.Y.; Liu, D.J.; Yang, X.M.; Wang, Y.H.; Yang, M.W.; Fu, X.L.; Zhang, X.X.; et al. Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 153, 277–291 e19. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apelin | APJ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics | N° of Cases N = 49 | Negative | Weak | Medium | Strong | p-Value | Negative | Weak | Medium | Strong | p-Value |
| Age (years) | 0.24 | 0.58 | |||||||||
| < 60 | 9 (18.4%) | 0 (0%) | 5 (56%) | 4 (44%) | 0 (0%) | 2 (22.2%) | 5 (55.6%) | 1 (11.1%) | 1 (11.1%) | ||
| ≥ 60 | 40 (81.6%) | 1 (2.5%) | 17 (42.5%) | 10 (25%) | 12 (30%) | 7 (17.5%) | 16 (40%) | 16 (40%) | 1 (2.5%) | ||
| Gender | 0.72 | 0.56 | |||||||||
| Female | 27 (55.1%) | 0 (0%) | 12 (44.4%) | 9 (33.3%) | 6 (22.3%) | 5 (18.5%) | 13 (48.2%) | 8 (29.6%) | 1 (3.7%) | ||
| Male | 22 (44.9%) | 1 (4.5%) | 10 (45.5%) | 5 (22.7%) | 6 (27.3%) | 4 (18.2%) | 9 (40.9%) | 8 (36.4%) | 1 (4.5%) | ||
| Adenocarcinoma | 49 | 1 (2%) | 22 (44.9%) | 14 (28.6%) | 12 (24.5%) | 9 (18.3%) | 21 (42.9%) | 17 (34.7%) | 2 (4.1%) | ||
| Histopathologic grade | 0.84 | 0.46 | |||||||||
| WD | 27 (55.1%) | 1(3.7%) | 12 (44.5%) | 8 (29.6%) | 6 (22.2%) | 6 (22.2%) | 14 (51.9%) | 6 (22.2%) | 1 (3.70%) | ||
| MD | 14 (28.6%) | 0 (0%) | 6 (42.9%) | 5 (35.7%) | 3 (21.4%) | 2 (14.3%) | 5 (35.7%) | 6 (42.9%) | 1 (7.1%) | ||
| PD | 8 (16.3%) | 0 (0%) | 5 (62.5%) | 1 (12.5%) | 2 (25%) | 1 (12.5%) | 2 (25%) | 5 (62.5%) | 0 (0%) | ||
| TNM stage | |||||||||||
| T1 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0.74 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0.98 |
| T2 | 8 (16.3%) | 0 (0%) | 3 (37.5%) | 2 (25%) | 3 (37.5%) | 2 (25%) | 4 (50%) | 2 (25%) | 0 (0%) | ||
| T3 | 40 (81.6%) | 1 (2.5%) | 19 (47.5%) | 11 (27.5%) | 9 (22.5%) | 7 (17.5%) | 16 (40%) | 15 (37.5%) | 2 (5%) | ||
| T4 | 1 (2%) | 0 (0%) | 0 (0%) | 1 (100%) | 0 (0%) | 0 (0%) | 1 (100%) | 0 (0%) | 0 (0%) | ||
| N0 | 13 (26.5%) | 1 (7.7%) | 6 (46.2%) | 4 (30.8%) | 2 (15.3%) | 0.33 | 3 (23.1%) | 7 (53.8%) | 2 (15.4%) | 1 (7.7%) | 0.36 |
| N1 | 36 (73.5%) | 0 (0%) | 16 (44.4%) | 10 (27.8%) | 10 (27.8%) | 6 (16.6%) | 14 (38.9%) | 15 (41.7%) | 1 (2.8%) | ||
| PanINs | 0.64 | 0.65 | |||||||||
| Negative | 7 | 7 (100%) | 7 (100%) | ||||||||
| PanIN 1 | 3 | 0 (0%) | 3 (100%) | 0 (0%) | 0 (0%) | 1 (33.3%) | 1 (33.3%) | 1 (33.3%) | 0 (0%) | ||
| PanIN 2 | 18 | 1 (5.5%) | 5 (27.8%) | 9 (50%) | 3 (16.7%) | 1 (5.6%) | 10 (55.6%) | 5 (27.8%) | 2 (11%) | ||
| PanIN 3 | 30 | 0 (0%) | 14 (46.6%) | 8(26.7%) | 8 (26.7%) | 3 (10%) | 18 (60%) | 7 (23.3%) | 2 (6.7%) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaves-Almagro, C.; Auriau, J.; Dortignac, A.; Clerc, P.; Lulka, H.; Deleruyelle, S.; Projetti, F.; Nakhlé, J.; Frances, A.; Berta, J.; et al. Upregulated Apelin Signaling in Pancreatic Cancer Activates Oncogenic Signaling Pathways to Promote Tumor Development. Int. J. Mol. Sci. 2022, 23, 10600. https://doi.org/10.3390/ijms231810600
Chaves-Almagro C, Auriau J, Dortignac A, Clerc P, Lulka H, Deleruyelle S, Projetti F, Nakhlé J, Frances A, Berta J, et al. Upregulated Apelin Signaling in Pancreatic Cancer Activates Oncogenic Signaling Pathways to Promote Tumor Development. International Journal of Molecular Sciences. 2022; 23(18):10600. https://doi.org/10.3390/ijms231810600
Chicago/Turabian StyleChaves-Almagro, Carline, Johanna Auriau, Alizée Dortignac, Pascal Clerc, Hubert Lulka, Simon Deleruyelle, Fabrice Projetti, Jessica Nakhlé, Audrey Frances, Judit Berta, and et al. 2022. "Upregulated Apelin Signaling in Pancreatic Cancer Activates Oncogenic Signaling Pathways to Promote Tumor Development" International Journal of Molecular Sciences 23, no. 18: 10600. https://doi.org/10.3390/ijms231810600
APA StyleChaves-Almagro, C., Auriau, J., Dortignac, A., Clerc, P., Lulka, H., Deleruyelle, S., Projetti, F., Nakhlé, J., Frances, A., Berta, J., Gigoux, V., Fourmy, D., Dufresne, M., Gomez-Brouchet, A., Guillermet-Guibert, J., Cordelier, P., Knibiehler, B., Jockers, R., Valet, P., ... Masri, B. (2022). Upregulated Apelin Signaling in Pancreatic Cancer Activates Oncogenic Signaling Pathways to Promote Tumor Development. International Journal of Molecular Sciences, 23(18), 10600. https://doi.org/10.3390/ijms231810600