
Intercontinental Gut Microbiome Variances in IBD
 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Participants’ Characteristics and Fecal Samples
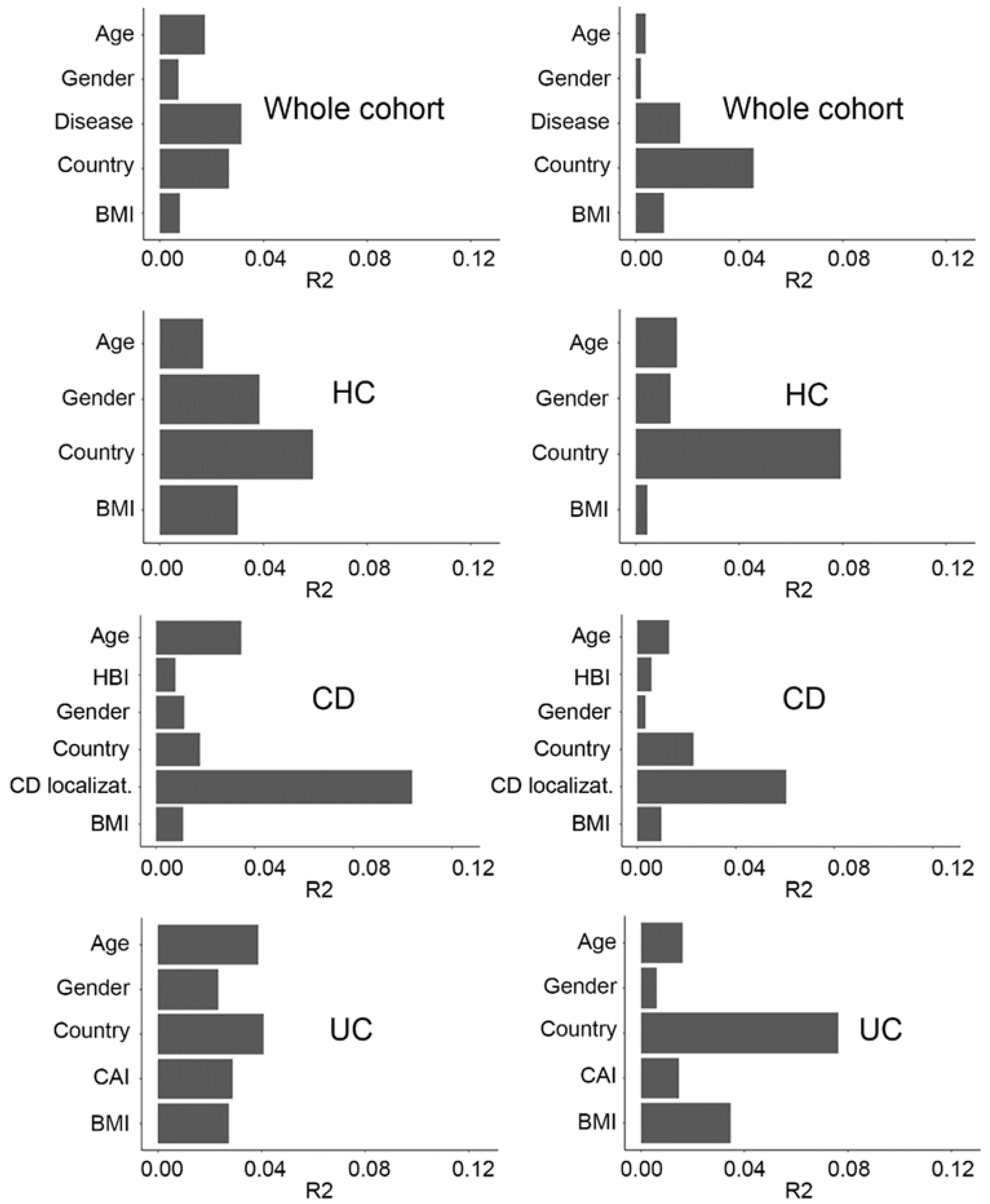
2.2. Fecal Microbiome Variation Explained by Covariates in the Metadata
2.3. Geographical and Disease-Related Dysbiosis
2.4. Taxonomic Profile Associated with Disease Phenotype and Severity
2.5. Taxonomic Differences across Countries and Health Status
3. Discussion
4. Materials and Methods
4.1. Collection of Metagenomic Data
4.2. Filtering of the Datasets
4.3. Upstream Sequence Analysis: Quality Control, Decontamination and Profiling
4.4. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bernstein, C.N.; Forbes, J.D. Gut Microbiome in Inflammatory Bowel Disease and Other Chronic Immune-Mediated Inflammatory Diseases. Inflamm. Intest. Dis. 2017, 2, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Seksik, P.; Rigottier-Gois, L.; Gramet, G.; Sutren, M.; Pochart, P.; Marteau, P.; Jian, R.; Doré, J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 2003, 52, 237–242. [Google Scholar] [CrossRef]
- Sheehan, D.; Shanahan, F. The Gut Microbiota in Inflammatory Bowel Disease. Gastroenterol. Clin. North Am. 2017, 46, 143–154. [Google Scholar] [CrossRef]
- Zuo, T.; Kamm, M.A.; Colombel, J.-F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef]
- Copeland, J.K.; Chao, G.; Vanderhout, S.; Acton, E.; Wang, P.W.; Benchimol, E.I.; El-Sohemy, A.; Croitoru, K.; Gommerman, J.L.; Guttman, D.S.; et al. The Impact of Migration on the Gut Metagenome of South Asian Canadians. Gut Microbes 2021, 13, 1–29. [Google Scholar] [CrossRef]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e10. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin Study Indicates Loss of Interaction Between Microbiota and Mucosa of Patients with Ulcerative Colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Metwaly, A.; Reitmeier, S.; Haller, D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 383–397. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Yilmaz, B.; Juillerat, P.; Øyås, O.; Ramon, C.; Bravo, F.D.; Franc, Y.; Fournier, N.; Michetti, P.; Mueller, C.; Geuking, M.; et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med. 2019, 25, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chang, Y.; Zhang, K.; Chen, H.; Tao, S.; Zhang, Z. Implication of the gut microbiome composition of type 2 diabetic patients from northern China. Sci. Rep. 2020, 10, 5450. [Google Scholar] [CrossRef]
- Reitmeier, S.; Kiessling, S.; Clavel, T.; List, M.; Almeida, E.L.; Ghosh, T.S.; Neuhaus, K.; Grallert, H.; Linseisen, J.; Skurk, T.; et al. Arrhythmic Gut Microbiome Signatures Predict Risk of Type 2 Diabetes. Cell Host Microbe 2020, 28, 258–272.e6. [Google Scholar] [CrossRef]
- Langhorst, J.; Schöls, M.; Cinar, Z.; Eilert, R.; Kofink, K.; Paul, A.; Zempel, C.; Elsenbruch, S.; Lauche, R.; Ahmed, M.; et al. Comprehensive Lifestyle-Modification in Patients with Ulcerative Colitis–A Randomized Controlled Trial. J. Clin. Med. 2020, 9, 3087. [Google Scholar] [CrossRef]
- Relling, I.; Akcay, G.; Fangmann, D.; Knappe, C.; Schulte, D.M.; Hartmann, K.; Müller, N.; Türk, K.; Dempfle, A.; Franke, A.; et al. Role of wnt5a in Metabolic Inflammation in Humans. J. Clin. Endocrinol. Metab. 2018, 103, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Eckenberger, J.; Laserna-Mendieta, E.; Sexton, K.A.; Bernstein, M.T.; Vagianos, K.; Sargent, M.; Ryan, F.; Moran, C.; Sheehan, D.; et al. Ranking microbiome variance in inflammatory bowel disease: A large longitudinal intercontinental study. Gut 2021, 70, 499–510. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gómez, G.; Mayorga, L.; Oyarzun, I.; Roca, J.; Borruel, N.; Casellas, F.; Varela, E.; Pozuelo, M.; Machiels, K.; Guarner, F.; et al. Dysbiosis and relapse-related microbiome in inflammatory bowel disease: A shotgun metagenomic approach. Comput. Struct. Biotechnol. J. 2021, 19, 6481–6489. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Gao, Y.; Jie, Z.; Yu, X.; Laursen, J.M.; Xiao, L.; Li, Y.; Li, L.; Zhang, F.; Feng, Q.; et al. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. Gigascience 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication; Wiley Online Library: Hoboken, NJ, USA, 1948. [Google Scholar]
- Martinez, I.; Stegen, J.C.; Maldonado-Gomez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The Gut Microbiota of Rural Papua New Guineans: Composition, Diversity Patterns, and Ecological Processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef]
- He, Y.; Wu, W.; Zheng, H.M.; Li, P.; McDonald, D.; Sheng, H.F.; Chen, M.X.; Chen, Z.H.; Ji, J.Y.; Zheng, Z.D.X.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Shanahan, F.; O’Mahony, C.; Marchesi, J.R. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J. Clin. Microbiol. 2006, 44, 3980–3988. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.-F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Monk, J.M.; Nurk, S.; Akseshina, M.; Zhu, Q.; Gemmell, C.; Gianetto-Hill, C.; Leung, N.; Szubin, R.; Sanders, J.; et al. Metagenomics-Based, Strain-Level Analysis of Escherichia coli from a Time-Series of Microbiome Samples from a Crohn’s Disease Patient. Front Microbiol. 2018, 9, 2559. [Google Scholar] [CrossRef] [PubMed]
- Companys, J.; Gosalbes, M.; Pla-Pagà, L.; Calderón-Pérez, L.; Llauradó, E.; Pedret, A.; Valls, R.; Jiménez-Hernández, N.; Sandoval-Ramirez, B.; del Bas, J.; et al. Gut Microbiota Profile and Its Association with Clinical Variables and Dietary Intake in Overweight/Obese and Lean Subjects: A Cross-Sectional Study. Nutrients 2021, 13, 2032. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arango, L.F.; Barrett, H.L.; McIntyre, H.D.; Callaway, L.K.; Morrison, M.; Nitert, M.D.; SPRING Trial Group. Connections Between the Gut Microbiome and Metabolic Hormones in Early Pregnancy in Overweight and Obese Women. Diabetes 2016, 65, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Brahe, L.K.; Le Chatelier, E.; Prifti, E.; Pons, N.; Kennedy, S.B.; Hansen, T.; Pedersen, O.; Astrup, A.; Ehrlich, S.; Larsen, L.H. Specific gut microbiota features and metabolic markers in postmenopausal women with obesity. Nutr. Diabetes 2015, 5, e159. [Google Scholar] [CrossRef]
- Valles-Colomer, M.; Darzi, Y.; Vieira-Silva, S.; Falony, G.; Raes, J.; Joossens, M. Meta-omics in Inflammatory Bowel Disease Research: Applications, Challenges, and Guidelines. J. Crohn’s Colitis 2016, 10, 735–746. [Google Scholar] [CrossRef]
- Lewis, J.D.; Abreu, M.T. Diet as a Trigger or Therapy for Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 398–414.e6. [Google Scholar] [CrossRef]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database C. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Silverberg, M.S.; Satsangi, J.; Ahmad, T.; Arnott, I.D.R.; Bernstein, C.N.; Brant, S.R.; Caprilli, R.; Colombel, J.-F.; Gasche, C.; Geboes, K.; et al. Toward an Integrated Clinical, Molecular and Serological Classification of Inflammatory Bowel Disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. Gastroenterol. 2005, 19, 5A–36A. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.F.; Bradshaw, J.M. A simple index of Crohn’s-disease activity. Lancet 1980, 1, 514. [Google Scholar] [CrossRef]
- Walmsley, R.S.; Ayres, R.C.S.; Pounder, R.E.; Allan, R.N. A simple clinical colitis activity index. Gut 1998, 43, 29–32. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| United States | Spain | China | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type of Cohort | CD | UC | Non-IBD | Total | CD | UC | Non-IBD | Total | CD | UC | Non-IBD | Total |
| Number of subjects | 37 | 22 | 20 | 79 | 33 | 33 | 67 | 133 | 62 | NA | 52 | 114 |
| Number of samples | 506 | 319 | 342 | 1167 | 57 | 65 | 67 | 189 | 62 | NA | 52 | 114 |
| CD location (n) L1/L1+L4 L2/L2+L4 L3/L3+L4 | 109/45 58/23 128/143 | NA | NA | NA | 18/2 0/0 35/2 | NA | NA | NA | NA | NA | NA | NA |
| Gender (% female) | 39.3 | 65.8 | 50.9 | 50.0 | 66.7 | 72.3 | 56.7 | 65.1 | 25.8 | NA | 5.8 | 5.8 |
| Age (mean ± SD) | 22.5 ± 14.2 | 29.2 ± 19.5 | 27.5 ± 19.5 | 27.1 ± 17.7 | 35.8 ± 11.8 | 40.1 ± 10.5 | 43.1 ± 15.9 | 27.5±16.9 | 28.4 ± 8.1 | NA | 20.5 ± 7.8 | 20.5 ± 7.8 |
| BMI (mean ± SD) | 23.3 ± 7.5 | 23.0 ± 6.8 | 23.4 ± 7.0 | 23.3 ± 6.8 | 22.4 ± 4.0 | 23.6 ± 4.1 | 25.0 ± 4.3 | 23.2±6.7 | 18.6 ± 2.8 | NA | 21.4 ± 3.3 | 21.4 ± 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayorga, L.; Serrano-Gómez, G.; Xie, Z.; Borruel, N.; Manichanh, C. Intercontinental Gut Microbiome Variances in IBD. Int. J. Mol. Sci. 2022, 23, 10868. https://doi.org/10.3390/ijms231810868
Mayorga L, Serrano-Gómez G, Xie Z, Borruel N, Manichanh C. Intercontinental Gut Microbiome Variances in IBD. International Journal of Molecular Sciences. 2022; 23(18):10868. https://doi.org/10.3390/ijms231810868
Chicago/Turabian StyleMayorga, Luis, Gerard Serrano-Gómez, Zixuan Xie, Natalia Borruel, and Chaysavanh Manichanh. 2022. "Intercontinental Gut Microbiome Variances in IBD" International Journal of Molecular Sciences 23, no. 18: 10868. https://doi.org/10.3390/ijms231810868
APA StyleMayorga, L., Serrano-Gómez, G., Xie, Z., Borruel, N., & Manichanh, C. (2022). Intercontinental Gut Microbiome Variances in IBD. International Journal of Molecular Sciences, 23(18), 10868. https://doi.org/10.3390/ijms231810868