1. Introduction
Infection is one of the prominent causes of human morbidity and mortality, especially in patients requiring critical care [
1,
2]. Moreover, in intensive care units (ICUs), a serious complication of infection is sepsis and its maximal manifestation, septic shock [
3]. Sepsis is an infection-induced life-threatening organ dysfunction with mortality rates reaching 20–70% [
4,
5].
Infectious diseases have been found to be a major selective pressure [
6]. Despite the ambiguity of the precise etiology of sepsis, numerous studies have shown that gene polymorphisms have an important role in affecting individual susceptibility to sepsis [
7]. Some polymorphisms of the innate immune system are supposed to mediate a predisposition to infectious complications including the outcome of patients with sepsis [
8]. The innate immune system is of crucial importance for both the direct defense against micro-organisms and the activation of the adaptive immune system [
9]. The innate immunity system is the main mediator of inflammation, and it recruits specific pattern recognition receptors (PRRs) capable of recognizing micro-organisms through identifying conserved pathogen-associated molecular patterns.
Toll-like receptors (TLR) are the most studied subtypes of pattern recognition receptors with their critical importance in the immune system [
10,
11]. Among the members of the TLR family, TLR2 and TLR4 are considered the most important PRRs that cover a wide range of antigenic determinants [
12]. TLR4 has a distinctive ability to recognize a very wide range of microorganisms including Gram-negative bacteria through Lipopolysaccharide (LPS), in addition to many viruses and Fungi. Meanwhile, TLR2 is regarded as a key molecule in regulating our immune system with a crucial role in the recognition of Peptidoglycans of Gram-positive bacteria, in addition to different ligands of yeast, fungi, viruses, and parasites [
12,
13].
One of the most studied innate immunity polymorphisms is the
TLR4 Asp299Gly (rs4986790) polymorphism, which interferes with TLR4 signal transduction; thus, it is supposed to affect host susceptibility to infections and microbial invasions [
14]. Moreover, structural analysis of
TLR4 Asp299Gly has revealed evidence of a resulted impairment in TLR4 binding to its ligands [
15]. Meanwhile, one of the most important polymorphisms of
TLR2 is Arg753Gln; the presence of this SNP was found to impair the signaling pathway of this key receptor [
16], thus suggesting increased susceptibility to infections and sepsis.
Consequently, many studies have been conducted all over the world to reveal the prevalence of these SNPs and their impact on infection and sepsis susceptibility, but a varied pattern of prevalence was found for both
TLR4 and
TLR2 SNPs among different populations [
13,
17,
18]. In addition, conflicting results were found regarding their impact on infection and sepsis susceptibility in different populations [
19,
20,
21]. Therefore, a need was felt for further investigation on these issues. The usage of computational approaches in studying SNPs’ impact has gained momentum and importance in recent years [
22,
23,
24,
25]. Integrating the in silico approach with the experimental one provides great accuracy and depth to the analysis.
In this study, we aimed to investigate the possible role of TLR2 and TLR4 polymorphisms in affecting sepsis susceptibility and survival in critically ill patients in the Egyptian population using both in silico analysis and experimental methods.
3. Discussion
The remarkable importance of TLR2 and TLR4 in our immune system and in modulating our response to infection suggested potential roles of their important variants, Arg753Gln and Asp299Gly, in increasing susceptibility to infection and sepsis as well.
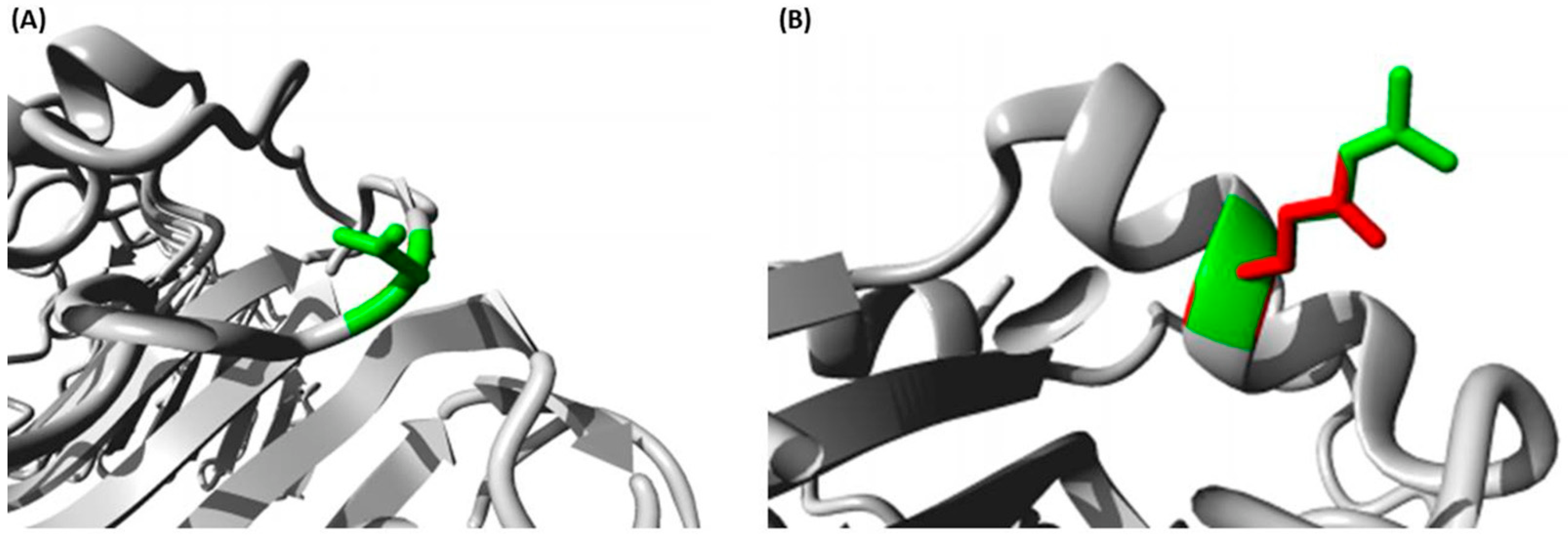
Different bioinformatics approaches were utilized in our analysis. Investigating the impacts of our variants depended on five various tools with various approaches to achieve a high robustness and effectiveness. While rs4986790 was predicted to possess a benign impact on TLR4 by all tools, rs5743708 was predicted by all tools except SNPs and GO to possess a damaging impact on TLR2. Moreover, the SNPs’ positions on the domains of their proteins were determined by InterPro, revealing the presence of rs5743708 on the important TIR domain. The TIR domain has a crucial role in the activation of TLR pathways [
26]. Therefore, it is anticipated that this mutation could affect its protein function. In addition, since protein function and structure are critically dependent on its stability [
27], the impacts of rs5743708 and rs4986790 on their proteins’ stability were investigated revealing how proteins’ stability was reduced by these SNPs. Furthermore, concerning the relationship between high scores of conservation and functionally significant residues [
28], the conservation analysis was intended to anticipate those SNPs which could affect the significant functions. Rs5743708 of
TLR2 was found to be a functional residue with high conservation. On the contrary, rs4986790 of
TLR4 was found to be a variable residue. In addition, both rs5743708 and rs4986790 were anticipated to induce structural impacts on TLR2 and TLR4, respectively using the HOPE bioinformatics server.
In our prospective study, the genotype frequencies for
TLR4 were in accordance with Hardy–Weinberg equilibrium. On the contrary, the genotype frequencies for
TLR2 were not in accordance with the Hardy–Weinberg equilibrium, and this aberrant result was also found by Saleh et al. in the Egyptian population, in his study about Toll-like receptor-2 polymorphisms and the susceptibility to pulmonary and peritoneal tuberculosis [
29] which may require further investigation. The different prevalence of these SNPs between different populations have been steadily observed by different researchers [
13,
17,
18] with obvious differences between Asian, African, and European ethnicities for both SNPs. These different distribution patterns between populations were suspected to be responsible for different susceptibility patterns to infectious diseases and other serious diseases such as coronary artery disease and type 2 Diabetes as well [
18,
30].
In our study, there was a significant association between TLR2 Arg753Gln polymorphism and sepsis under the over-dominant model (
p = 0.043), while the TLR4 polymorphism did not show such significance. Some other investigators reached the same results in some populations despite the observed conflict between studies. A meta-analysis study conducted by Gao and colleagues found an association in this study between Arg753Gln SNP and the risk of sepsis among critically ill adult patients in Europe. Meanwhile, this study also shed light on the issue of the conflicting results regarding the TLR2 polymorphism and developing sepsis [
21]. The TLR4 polymorphism studies also showed conflicting results; a study conducted in France by Lorenz et al. found that the Asp299Gly and Thr399Ile polymorphisms of TLR4 might potentially be linked to Gram-negative septic shock [
19]. On the contrary, some studies showed an absence of association between TLR4 SNP and sepsis; a study conducted by Kumpf et al. found no association between Asp299Gly and Thr399Ile polymorphisms of TLR4 and the incidence of sepsis syndrome or the type of organisms causing surgical infection in German adults [
20]. In addition, another study by Shan Xo et al. in Wenzhou found that the Asp299Gly and Thr399Ile polymorphisms may not correlate with susceptibility to sepsis in Chinese Han children [
31]. These conflicting results can be seen frequently among different ethnic groups in these types of genetic association studies investigating diseases that depend on several genetic factors [
32], as sepsis is believed to be initiated and augmented by multiple genes and there is no full control over sepsis by a single gene [
33,
34]. Consequently, the various frequency of different SNPs in different ethnic groups, and the difference in the penetration and the effect of SNPs because of other factors such as gender or age variations in different studies could explain these conflicting results among different populations.
Developing infection with
Acinetobacter baumannii was found to have a statistically significant association with the TLR4 polymorphism (
p = 0.001). This finding is in agreement with a recent study conducted by Chatzi et al. who found that the Asp299Gly and Thr399Ile polymorphisms of TLR4 could play an essential role in developing multidrug resistance to
Acinetobacter baumannii in CNS infections [
35]. In addition, other researchers have confirmed the role of TLR4 in
Acinetobacter baumannii infection in vitro and in vivo and found that the production of IL-8 by epithelial A549 cells in the human lung as a response to
Acinetobacter baumannii required both TLR2 and TLR4 [
36]. However, other studies showed that the recognition of
Acinetobacter baumannii depends on TLR4 rather than TLR2, as TLR4 is the dominant receptor in this type of recognition. Knapp et al. found that TLR4-deficient mice, not TLR2-deficient mice (with intranasal inoculation of
Acinetobacter baumannii Lipopolysaccharides) showed the impaired production of TNFa in bronchi alveolar lavage fluid and the impaired recruitment of polymorph nuclear cells, compared with Wild Type mice [
37]. Moreover, Kim et al. found that the production of
Acinetobacter baumannii-induced cytokines was impaired with TLR4-deficient bone marrow-derived macrophages or dendritic cells, while it was not the case with TLR2-deficient macrophages [
38]. Besides, Erridge et al. found that the activation of human monocytes (resulting from phenol water re-extracted Lipopolysaccharides from
Acinetobacter baumannii) was the responsibility of the TLR4 signaling pathway [
39]. This association between the TLR4 polymorphism and this virulent bacterium could allow proper management and prevention measures where high rates of
Acinetobacter baumannii infection are found. This could be an important step towards the individualization of host susceptibilities towards virulent microorganisms in intensive care units.
Our study also found a significant association between the
TLR4 polymorphism (rs4986790) and the post-surgical category among patients referred to ICU. This role of the TLR4 polymorphism in post-surgery was investigated by a clinical study conducted by Koch and colleagues who found that the presence of a
TLR4 polymorphism influenced the immune–endocrine stress response which resulted from the systemic inflammation caused by major surgery. They found decreased serum concentrations of ACTH, IL-8, IL-10, and GM-CSF postoperatively in those surgical patients who carried that polymorphism [
40]. This might explain this significant association found in our study.
The multivariate analysis was also performed to analyze the effects of our variables on the development of sepsis syndrome, but it was only the age factor that was found to have an independent association with the risk of sepsis in our study group. The age factor is a well-identified risk factor for developing this syndrome [
41].
Survival analysis found that the length of stay and the surgical category of admission had a significant association with time of survival in intensive care units. Our results are in agreement with several studies that found an association between prolonged ICU stay and higher hospital mortality as well. Those patients, with an ICU length of stay of 14 days or longer, were found to have a mortality rate of more than 50% [
42,
43].
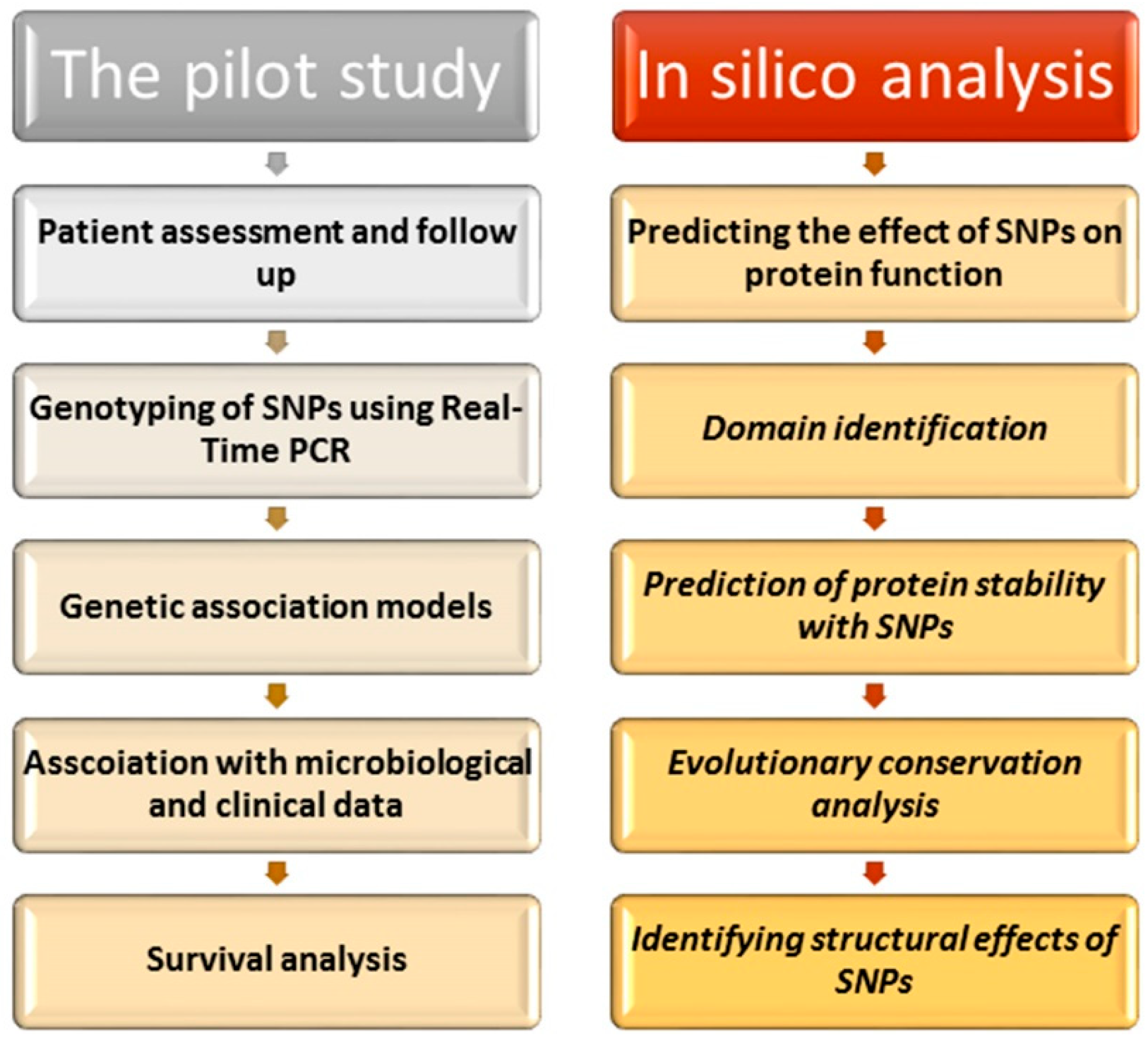
Overall, our study is characterized by the usage of both experimental and in silico methods. Our investigation showed promising results regarding the analysis of the role of TLRs variants in infection and sepsis. However, our study had its limitations as in most genetic polymorphism studies, as the number of patients carrying the variant alleles was relatively small due to the small number of these polymorphisms in the general population. As a result, there is a need for multi-center studies conducted on a larger scale to validate these findings.
4. Materials and Methods
4.1. Ethics Statement
The study protocol was approved by Scientific Research Ethics Commission at Suez Canal University (reference No. 201709MH1). All subjects or their next of kin gave informed consent before inclusion in the study.
4.2. In Silico Analysis
4.2.1. General Information
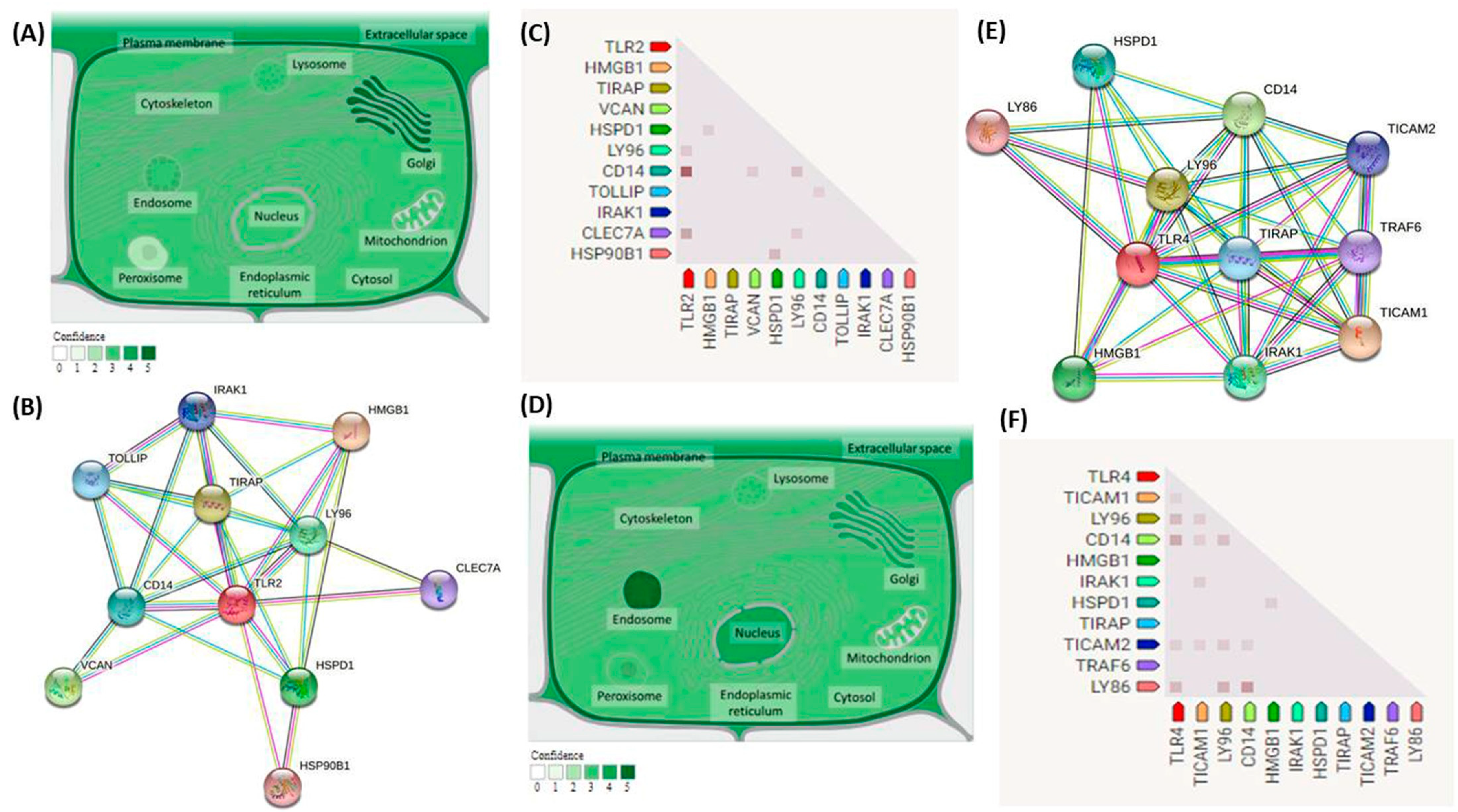
National Center for Biotechnology Information (NCBI) and Ensembl databases were used to retrieve general information about
TLR2 and
TLR4 genes. Subcellular localization was retrieved from
compartments.jensenlab.org mainly and
genecards.org. Gene coexpression and predicted protein–protein interactions were obtained from the String Biological database. General information about rs5743708 and rs4986790 were brought from the dbSNP and Ensembl databases. (
https://web.expasy.org (accessed on 29 August 2021)) was used for retrieving data about the variants’ effect on sequences of our proteins with these data gained from UniProtKB/Swiss-Prot databases.
4.2.2. Predicting the Effect of SNPs on Protein Function
Five bioinformatics tools were used to predict the effect of SNPs on protein function to increase the strength and accuracy of results; 1-SIFT (Sorting Intolerant from Tolerant) (
https://sift.bii.a-star.edu.sg/ (accessed on 30 August 2021)). SIFT depends on sequence homology in addition to the physical properties of amino acids to predict the effect of missense mutations on protein function [
44]. 2-PolyPhen-2 (Polymorphism Phenotyping v2) (
http://genetics.bwh.harvard.edu/pph2 (accessed on 30 August 2021)). PolyPhen-2 uses comparative and physical approaches to predict the effect of amino acid substitution [
45]. 3-PANTHER (Protein Analysis Trough Evolutionary Relationship) (
http://www.pantherdb.org/tools/csnpScoreForm.jsp (accessed on 30 August 2021)). This method depends on calculating the evolutionary preservation of an amino acid to predict the likelihood that a nonsynonymous SNP could cause a functional impact on the protein [
46]. 4- PROVEAN (Protein Variation Effect Analyzer) (
http://provean.jcvi.org/seq_submit.php (accessed on 30 August 2021)). PROVEAN uses blast hits to calculate the delta alignment score and computes the PROVEAN score finally with a cutoff at −2.5 [
47]. 5-SNPs and GO (
https://snps.biofold.org/snps-and-go/snps-and-go.html (accessed on 30 August 2021)). SNPs and GO depend on protein functional annotation to predict the impact of variations [
48].
4.2.3. The Identification of SNP Location on Protein Domains
The locations of SNPs on conserved domains on TLR2 and TLR4 proteins were identified using the InterPro bioinformatics tool (
https://www.ebi.ac.uk/interpro/ (accessed on 30 August 2021)), a bioinformatics tool that could perform functional analysis of protein and identify domains and functional sites [
49].
4.2.4. The Prediction of Protein Stability with SNPs
We used I-Mutant 2.0 (
https://folding.biofold.org/i-mutant/i-mutant2.0.html (accessed on 30 August 2021)) to predict the stability of the TLR2 and TLR4 proteins with rs5743708 and rs4986790 SNPs, respectively [
50]. I-Mutant 2.0 is considered a support vector machine that was tested depending on the ProTherm database which contained the largest experimental data about stability changes with protein mutations [
51].
4.2.5. The Identification of Evolutionarily Conserved Positions in a Protein Sequence
This identification was performed using the ConSurf server (
https://consurf.tau.ac.il (accessed on 30 August 2021)) which depends on phylogenetic relations between homologous sequences to identify the evolutionary conservation of amino acids in protein sequences [
28,
52].
4.2.6. The Identification of Structural Effects of SNPs
Structural effects of rs5743708 and rs4986790 SNPs on TLR2 and TLR4, respectively, were analyzed using HOPE (
https://www3.cmbi.umcn.nl/hope/ (accessed on 30 August 2021)) which is a mutant analysis server that could analyze the effects of SNPs on protein structure [
53].
4.3. The Study Design
This was a prospective observational study that was conducted in intensive care units in Suez Canal University Hospitals, Ismailia, Egypt, for seven months. All ICU Patients who contracted infections with a positive culture or a chest X-ray were included in the study group. All included patients were Egyptian adults of both sexes. Exclusion criteria were patients younger than 18 years old, pregnancy, immune suppression, and patients with radiation therapy or chemotherapy.
Once admitted, general examination and clinical status were assessed for patients; both Acute Physiology and Chronic Health Evaluation (APACHE II) scores and sequential organ failure assessment (SOFA) scores were measured. In addition to vital signs check (blood pressure, heart rate, respiratory rate, central venous pressure, and temperature) and laboratory analyses such as complete blood count, blood sugar, CRP, blood urea nitrogen, serum calcium, potassium, sodium, aspartate aminotransferase, alanine aminotransferase, and arterial blood gas analysis were carried out.
The patients were further followed up to assess infection, sepsis, and septic shock. Routine cultures of sputum, blood, urine, and pus were collected to determine the presence of infection and identify the causing organism. Assessment of sepsis and septic shock was performed by daily evaluation for sepsis or septic shock. Sepsis and septic shock were defined and diagnosed according to “The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3)” [
4].
4.4. Samples Collection
Two milliliters of venous blood sample were collected into EDTA tubes from all admitted patients in the study group under complete aseptic conditions and stored at −80 °C until processed for DNA extraction.
4.5. Genotyping
Genomic DNA was extracted from venous blood with a QIAamp DNA Blood Mini kit (Cat. No. 51104; QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. The measurement of both the concentration and purity of the extracted DNA was performed by NanoDrop ND-1000 (NanoDrop Tech., Inc., Wilmington, DE, USA).
Genotyping for the TLR4 gene polymorphism (Asp299Gly; rs4986790) and TLR2 gene polymorphism (Arg753Gln; rs5743708) was performed using real-time polymerase chain reaction technology using TaqMan allelic discrimination assay. The required reagents for the TaqMan assay including TaqMan genotyping assay and TaqMan genotyping master mix were brought from Applied Biosystems (Foster City, CA, USA). The assay ID for rs5743708 is C_27860663_10 and for rs4986790 is C_11722238_20. PCR was run with a total reaction volume of 25 μL reaction volume. The components of PCR reaction were 12.5 μL TaqMan genotyping master mix; No AmpErase UNG (2×), genomic DNA (20 ng) diluted to 11.25 μL with DNase-RNase free water, and 1.25 μL TaqMan SNP genotyping assay mix (Cat. No. 4351379, Applied Biosystems, Foster City, USA). Nuclease-free water was used as a negative control.
The PCR amplification was carried out in a StepOne™ real-time PCR system (Applied Biosystems, Foster City, CA, USA) according to the following conditions: a hold cycle (95 °C for 10 min) followed by a 40-cycle PCR consisting of 95 °C for 15 s and 60 °C for one minute. SDS software version 1.3.1 (Applied Biosystems) was used for allelic discrimination. Genotyping was performed with blindness to sepsis/non-sepsis status.
4.6. Statistical Analysis
Statistical analysis was carried out using Microsoft
® Excel 2010 and the “Statistical Package for the Social Sciences (SPSS) for windows” software, version 24. Odds ratios (OR) with a 95% confidence interval (CI) were calculated. Descriptive statistics were expressed as percentages for qualitative variables and mean ± standard deviation (SD) for quantitative variables. Testing differences between septic patients and no septic patients were performed using Student’s
t-test, Chi-square (χ
2) test, or Fisher’s exact tests.
p-value was considered statistically significant below 0.05. The Hardy–Weinberg equilibrium (HWE) was calculated by the Online Encyclopedia for Genetic Epidemiology (OEGE) software (
http://www.oege.org/software/hwe-mrcalc.shtml (accessed on 10 March 2019)). The relationship between the risk factors including our polymorphisms and the development of sepsis was further determined using logistic regression after adjustment of factors. Survival analysis was performed as well. Log-rank, Breslow, and Tarone–Ware tests were used to find Kaplan–Meier estimates for survival. Cox regression analysis was applied to the data to determine if any of the variables were independently associated with the duration of survival.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}