
mRNA Capture Sequencing and RT-qPCR for the Detection of Pathognomonic, Novel, and Secondary Fusion Transcripts in FFPE Tissue: A Sarcoma Showcase

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
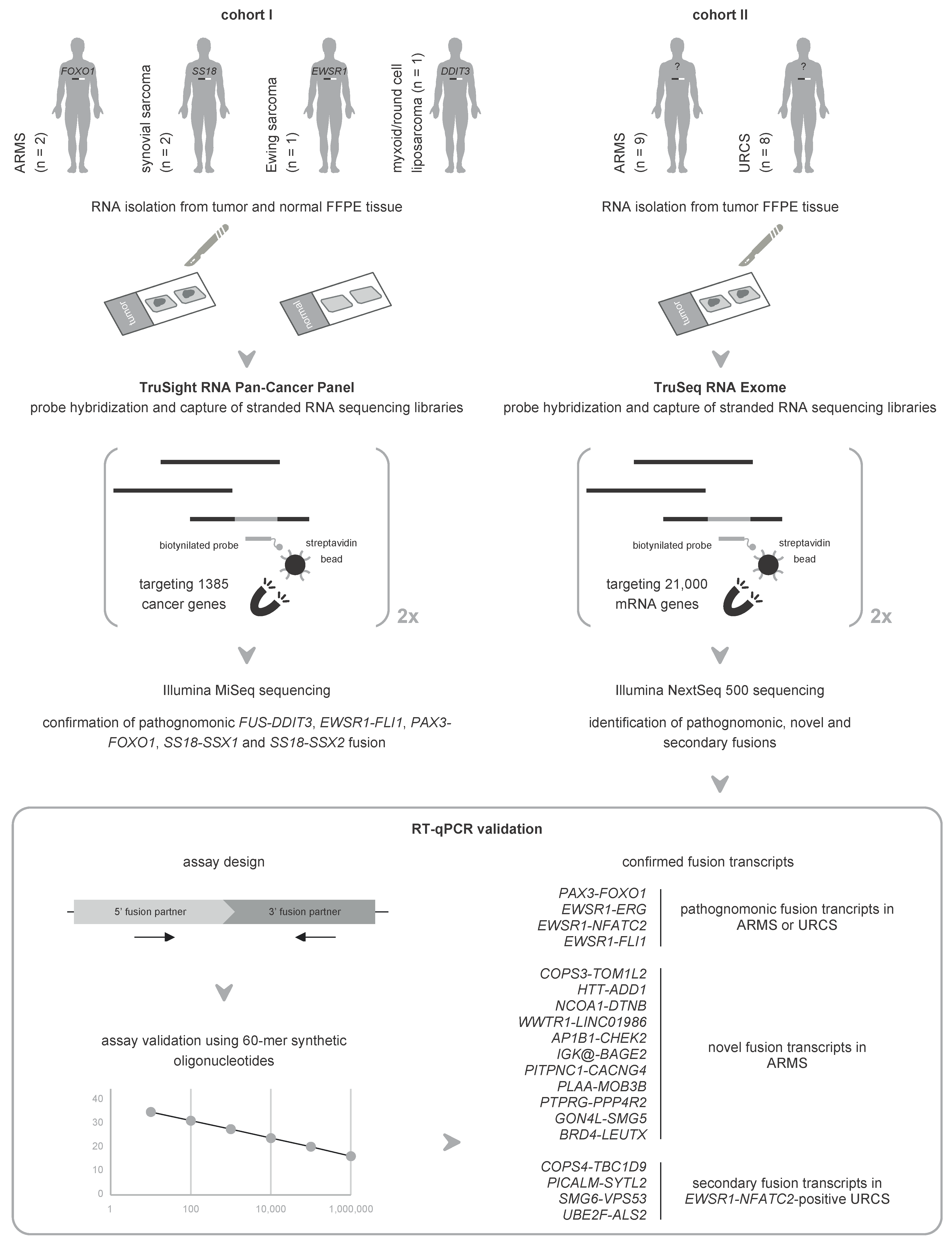
2.1. Orthogonal Validation of Known Fusions in Sarcoma Using mRNA Capture Sequencing
2.2. Unbiased mRNA Capture Sequencing Reveals Pathognomonic Fusion Transcripts in Clinicopathological Enigmatic Sarcomas
2.3. RT-qPCR Validates Pathognomonic Fusion Transcripts Detected by mRNA Capture Sequencing in FISH-Negative Sarcomas
2.4. Novel Fusion Transcripts Are Identified and Validated in Clinicopathological Enigmatic Sarcomas
2.5. RT-qPCR Confirms Recurrently Detected Secondary Fusion Transcripts in Sarcomas with an EWSR1-NFATC2 Fusion
3. Discussion
4. Materials and Methods
4.1. Patient Biomaterial
4.2. RNA Purification
4.3. mRNA Capture Sequencing
4.4. Fusion Transcript Identification and Selection for RT-qPCR Analysis
4.5. RT-qPCR Validation of Fusion Transcripts
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2021. Available online: https://mitelmandatabase.isb-cgc.org (accessed on 11 September 2022).
- Heyer, E.E.; Blackburn, J. Sequencing strategies for fusion gene detection. Bioessays 2020, 42, e2000016. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yeung, C.; Coleman, I.; Nelson, P.S.; Fang, M. Comparison of four next generation sequencing platforms for fusion detection: Oncomine by ThermoFisher, AmpliSeq by illumina, FusionPlex by ArcherDX, and QIAseq by QIAGEN. Cancer Genet. 2020, 243, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Velizheva, N.P.; Rechsteiner, M.P.; Valtcheva, N.; Freiberger, S.N.; Wong, C.E.; Vrugt, B.; Zhong, Q.; Wagner, U.; Moch, H.; Hillinger, S.; et al. Targeted next-generation-sequencing for reliable detection of targetable rearrangements in lung adenocarcinoma-a single center retrospective study. Pathol. Res. Pract. 2018, 214, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, D.; Brenca, M.; Baldazzi, D.; Goeman, F. Next-generation sequencing approaches for the identification of pathognomonic fusion transcripts in sarcomas: The experience of the Italian ACC Sarcoma Working Group. Front. Oncol. 2020, 10, 489. [Google Scholar] [CrossRef]
- Reeser, J.W.; Martin, D.; Miya, J.; Kautto, E.A.; Lyon, E.; Zhu, E.; Wing, M.R.; Smith, A.; Reeder, M.; Samorodnitsky, E.; et al. Validation of a targeted RNA sequencing assay for kinase fusion detection in solid tumors. J. Mol. Diagn. 2017, 19, 682–696. [Google Scholar] [CrossRef]
- Prieto-Conde, M.I.; Corchete, L.A.; García-álvarez, M.; Jiménez, C.; Medina, A.; Balanzategui, A.; Maldonado, R.; Sarasquete, M.E.; Alcoceba, M.; Puig, N.; et al. A new next-generation sequencing strategy for the simultaneous analysis of mutations and chromosomal rearrangements at DNA level in acute myeloid leukemia patients. J. Mol. Diagn. 2020, 22, 60–71. [Google Scholar] [CrossRef]
- Qadir, M.A.; Zhan, S.H.; Kwok, B.; Bruestle, J.; Drees, B.; Popescu, O.; Sorensen, P.H. ChildSeq-RNA: A next-generation sequencing-based diagnostic assay to identify known fusion transcripts in childhood sarcomas. J. Mol. Diagn. 2014, 16, 361–370. [Google Scholar] [CrossRef]
- Chung, J.; Son, D.; Jeon, H.; Kim, K.; Park, G. The minimal amount of starting DNA for Agilent’s hybrid capture-based targeted massively parallel sequencing. Sci. Rep. 2016, 6, 26732. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [PubMed]
- Heyer, E.E.; Deveson, I.W.; Wooi, D.; Selinger, C.I.; Lyons, R.J.; Hayes, V.M.; Toole, S.A.O.; Ballinger, M.L.; Gill, D.; Thomas, D.M.; et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat. Commun. 2019, 10, 1388. [Google Scholar] [PubMed]
- Levin, J.Z.; Berger, M.F.; Adiconis, X.; Rogov, P.; Melnikov, A.; Fennell, T.; Nusbaum, C.; Garraway, L.A.; Gnirke, A. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009, 10, R115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xia, J.; Jia, P.; Pao, W.; Zhao, Z. Application of next generation sequencing to human gene fusion detection: Computational tools, features and perspectives. Brief. Bioinform. 2013, 14, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef]
- Yoshida, A.; Arai, Y.; Kobayashi, E.; Yonemori, K.; Ogura, K.; Hama, N.; Mukai, W.; Motoi, T.; Kawai, A.; Shibata, T.; et al. CIC break-apart fluorescence in-situ hybridization misses a subset of CIC-DUX4 sarcomas: A clinicopathological and molecular study. Histopathology 2017, 71, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Nicorici, D.; Satalan, M.; Edgren, H.; Kangaspeska, S.; Murumagi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A Tool for Finding Somatic Fusion Genes in Paired-End RNA-Sequencing Data. bioRxiv 2014. [Google Scholar] [CrossRef]
- Kumar, S.; Vo, A.D.; Qin, F.; Li, H. Comparative assessment of methods for the fusion transcripts detection from RNA-seq data. Sci. Rep. 2016, 6, 21597. [Google Scholar] [CrossRef]
- Liu, S.; Tsai, W.; Ding, Y.; Chen, R.; Fang, Z.; Huo, Z.; Kim, S.; Ma, T.; Chang, T.; Priedigkeit, N.M.; et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016, 44, e47. [Google Scholar] [CrossRef]
- Patócs, B.; Németh, K.; Garami, M.; Arató, G.; Kovalszky, I.; Szendroi, M.; Fekete, G. Multiple splice variants of EWSR1-ETS fusion transcripts co-existing in the Ewing sarcoma family of tumors. Cell. Oncol. 2013, 36, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Zhao, X.; Patchefsky, A.S.; Flieder, D.B.; Talarchek, J.N.; Testa, J.R.; Wei, S. Clinical application of RNA sequencing in sarcoma diagnosis: An institutional experience. Medicine 2019, 98, e16031. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Lui, W.; Xie, Y.; Zhang, A.; Mandahl, N.; Larsson, C.; Larsson, O. Co-existence of SYT-SSX1 and SYT-SSX2 fusions in synovial sarcomas. Oncogene 2002, 21, 4181–4190. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Healey, J.H.; Zhang, L.; Meyers, P.; Tap, W.D.; Antonescu, C.R. The clinical heterogeneity of round cell sarcomas with EWSR1/FUS gene fusions: Impact of gene fusion type on clinical features and outcome. Genes Chromosomes Cancer 2020, 59, 525–534. [Google Scholar] [CrossRef]
- Perret, R.; Escuriol, J.; Velasco, V.; Mayeur, L.; Soubeyran, I.; Delfour, C.; Aubert, S.; Polivka, M.; Karanian, M.; Meurgey, A.; et al. NFATc2-rearranged sarcomas: Clinicopathologic, molecular, and cytogenetic study of 7 cases with evidence of AGGRECAN as a novel diagnostic marker. Mod. Pathol. 2020, 33, 1930–1944. [Google Scholar] [CrossRef]
- Diaz-Perez, J.A.; Nielsen, G.P.; Antonescu, C.; Taylor, M.S.; Lozano-Calderon, S.A.; Rosenberg, A.E. EWSR1/FUS-NFATc2 rearranged round cell sarcoma: Clinicopathological series of 4 cases and literature review. Hum. Pathol. 2019, 90, 45–53. [Google Scholar] [CrossRef]
- Yoshida, K.; Machado, I.; Motoi, T.; Kawai, A.; Antonescu, C.R.; Yoshida, A. NKX3-1 is a useful immunohistochemical marker of EWSR1-NFATC2 sarcoma and mesenchymal chondrosarcoma. Am. J. Surg. Pathol. 2020, 44, 719–728. [Google Scholar] [CrossRef]
- Creason, A.; Haan, D.; Dang, K.; Chiotti, K.E.; Inkman, M.; Lamb, A.; Yu, T.; Hu, Y.; Norman, T.C.; Buchanan, A.; et al. A community challenge to evaluate RNA-seq, fusion detection, and isoform quantification methods for cancer discovery. Cell Syst. 2021, 12, 827–838. [Google Scholar] [CrossRef]
- Yan, T.; Gokgoz, N.; Gill, M.; Eskandarian, S.; Parkes, R.K.; Bull, S.B.; Bell, R.S.; Andrulis, I.L. COPS3 amplification and clinical outcome in osteosarcoma. Cancer 2007, 109, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.; Aagesen, T.H.; Maelandsmo, G.M.; Lothe, R.A.; Myklebost, O.; Forus, A. Amplification and overexpression of COPS3 in osteosarcomas potentially target TP53 for proteasome-mediated degradation. Oncogene 2003, 22, 5358–5361. [Google Scholar] [CrossRef]
- Kao, Y.; Ranucci, V.; Zhang, L.; Sung, Y.; Athanasian, E.A.; Swanson, D.; Dickson, B.C.; Antonescu, C.R. Recurrent BRAF gene rearrangements in myxoinflammatory fibroblastic sarcomas, but not hemosiderotic fibrolipomatous tumors. Am. J. Surg. Pathol. 2017, 41, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, M.; Dettling, M.; Koscielniak, E.; Stegmaier, S.; Simon-Klingenstein, K.; Bu, P.; Niggli, F.K.; Scha, B.W. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004, 64, 5539–5545. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.M.; Sboner, A.; Zhang, L.; Kitabayashi, N.; Chen, C.; Sung, Y.S.; Wexler, L.H.; Laquaglia, M.P.; Edelman, M.; Sreekantaiah, C.; et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer 2013, 52, 538–550. [Google Scholar] [CrossRef]
- Mohamed, A.; Sun, C.; De Mello, V.; Selfe, J.; Missiaglia, E.; Shipley, J.; Murray, G.I.; Zammit, P.S.; Wackerhage, H. The Hippo effector TAZ (WWTR1) transforms myoblasts and TAZ abundance is associated with reduced survival in embryonal rhabdomyosarcoma. J. Pathol. 2016, 240, 3–14. [Google Scholar]
- Deel, M.D.; Slemmons, K.K.; Hinson, A.R.; Genadry, K.C.; Burgess, B.A.; Crose, L.E.S.; Kuprasertkul, N.; Oristian, K.M.; Bentley, R.C.; Linardic, C.M. The transcriptional coactivator TAZ is a potent mediator of alveolar rhabdomyosarcoma tumorigenesis. Clin. Cancer. Res. 2018, 24, 2616–2630. [Google Scholar]
- Fullenkamp, C.A.; Hall, S.L.; Jaber, O.I.; Pakalniskis, B.L.; Savage, C.; Savage, J.M.; Ofori-Amanfo, G.K.; Lambertz, A.M.; Ivins, S.D.; Stipp, C.S.; et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget 2016, 7, 30094–30108. [Google Scholar] [CrossRef]
- Mohamed, A.D.; Tremblay, A.M.; Murray, G.I.; Wackerhage, H. The Hippo signal transduction pathway in soft tissue sarcomas. Biochim. Biophys. Acta 2015, 1856, 121–129. [Google Scholar] [CrossRef]
- Doyle, L.A.; Fletcher, C.D.M.; Hornick, J.L. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am. J. Surg. Pathol. 2016, 40, 94–102. [Google Scholar]
- Lamar, J.M.; Nehru, V.M.; Weinberg, G. Epithelioid hemangioendothelioma as a model of YAP/TAZ-driven cancer: Insights from a rare fusion sarcoma. Cancers 2018, 10, 229. [Google Scholar] [CrossRef]
- Tanas, M.R.; Sboner, A.; Oliveira, A.M.; Erickson-Johnson, M.R.; Hespelt, J.; Hanwright, P.J.; Flanagan, J.; Luo, Y.; Fenwick, K.; Natrajan, R.; et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci. Transl. Med. 2011, 3, 98ra82. [Google Scholar] [CrossRef]
- Errani, C.; Zhang, L.; Sung, Y.S.; Hajdu, M.; Singer, S.; Maki, R.G. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 2011, 50, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, R.; Hergovich, A. MOB (Mps one binder) proteins in the Hippo pathway and cancer. Cells 2019, 8, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.D.; De Borja, R.; Young, M.D.; Fuligni, F.; Roberts, N.D.; Hajjar, S.; Layeghifard, M.; Novokmet, A.; Paul, E.; Anaka, M.; et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science 2018, 361, eaam8419. [Google Scholar] [CrossRef]
- Huang, S.-C.; Zhang, L.; Sung, Y.-S.; Chen, C.-L.; Kao, Y.-C.; Agaram, N.P.; Singer, S.; Tap, W.D.; D’Angelo, S.; Antonescu, C.R. Recurrent CIC gene abnormalities in angiosarcomas: A molecular study of 120 cases with concurrent investigation of PLCG1, KDR, MYC, and FLT4 gene alterations. Am. J. Surg. Pathol. 2016, 40, 645–655. [Google Scholar] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar]
- Barresi, S.; Giovannoni, I.; Rossi, S.; Stracuzzi, A.; Quacquarini, D.; Cafferata, B.; Piscitelli, D.; De Leonardis, F.; Marzullo, A.; Alaggio, R. A novel BRD4-LEUTX fusion in a pediatric sarcoma with epithelioid morphology and diffuse S100 expression. Genes Chromosomes Cancer 2021, 60, 647–652. [Google Scholar]
- Koelsche, C.; Kriegsmann, M.; Kommoss, F.K.F.; Stichel, D.; Kriegsmann, K.; Vokuhl, C.; Grünewald, T.G.P.; Romero-Pérez, L.; Kirchner, T.; de Alava, E.; et al. DNA methylation profiling distinguishes Ewing-like sarcoma with EWSR1-NFATc2 fusion from Ewing sarcoma. J. Cancer Res. Clin. Oncol. 2019, 145, 1273–1281. [Google Scholar]
- Watson, S.; Perrin, V.; Guillemot, D.; Reynaud, S.; Coindre, J.; Karanian, M.; Guinebretière, J.; Freneaux, P.; Le Loarer, F.; Bouvet, M. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J. Pathol. 2018, 245, 29–40. [Google Scholar] [CrossRef]
- Dupain, C.; Gracia, C.; Harttrampf, A.C.; Rivière, J.; Geoerger, B.; Massaad-massade, L. Newly identified LMO3-BORCS5 fusion oncogene in Ewing sarcoma at relapse is a driver of tumor progression. Oncogene 2019, 38, 7200–7215. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Song, Z.; Chang, M.; Song, Y.; Frierson, H.; Li, H. Recurrent cis-SAGe chimeric RNA, D2HGDH-GAL3ST2, in prostate cancer. Cancer Lett. 2016, 380, 39–46. [Google Scholar] [CrossRef]
- Chakravarthi, B.V.; Dedigama-Arachchige, P.; Carskadon, S.; Sundaram, S.K.; Li, J.; Wu, K.H.H.; Chandrashekar, D.S.; Peabody, J.O.; Stricker, H.; Hwang, C.; et al. Pseudogene associated recurrent gene fusion in prostate cancer. Neoplasia 2019, 21, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Xiao, Z.D.; Dai, Z.; Zhu, L.; Lei, H.; Diao, L.T.; Xiong, Y. The landscape of long noncoding RNA-involved and tumor-specific fusions across various cancers. Nucleic Acids Res. 2020, 48, 12618–12631. [Google Scholar] [CrossRef]
- Kangaspeska, S.; Hultsch, S.; Edgren, H.; Nicorici, D.; Murumägi, A.; Kallioniemi, O. Reanalysis of RNA-sequencing data reveals several additional fusion genes with multiple isoforms. PLoS ONE 2021, 7, e48745. [Google Scholar]
- Kloosterman, W.P.; Coebergh Van Den Braak, R.R.J.; Pieterse, M.; Van Roosmalen, M.J.; Sieuwerts, A.M.; Stangl, C.; Brunekreef, R.; Lalmahomed, Z.S.; Ooft, S.; Van Galen, A.; et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017, 77, 3814–3822. [Google Scholar] [CrossRef]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Kallioniemi, O. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12, R6. [Google Scholar] [CrossRef]
- Evaluating RNA Quality from FFPE Samples. Illumina Technical Note. 2021. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/evaluating-rna-quality-from-ffpe-samples-technical-note-470-2014-001.pdf (accessed on 2 May 2018).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Lefever, S.; Pattyn, F.; De Wilde, B.; Coppieters, F.; De Keulenaer, S.; Hellemans, J.; Vandesompele, J. High-throughput PCR assay design for targeted resequencing using primerXL. BMC Bioinform. 2017, 18, 400. [Google Scholar]
- Rihani, A.; Van Maerken, T.; Pattyn, F.; Van Peer, G.; Beckers, A.; De Brouwer, S.; Kumps, C.; Mets, E.; Van der Meulen, J.; Rondou, P.; et al. Effective Alu repeat based RT-qPCR normalization in cancer cell perturbation experiments. PLoS ONE 2013, 8, e71776. [Google Scholar]
- Ruijter, J.M.; Pfaffl, M.W.; Zhao, S.; Spiess, A.N.; Boggy, G.; Blom, J.; Rutledge, R.G.; Sisti, D.; Lievens, A.; De Preter, K.; et al. Evaluation of qPCR curve analysis methods for reliable biomarker discovery: Bias, resolution, precision, and implications. Methods 2013, 59, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Zeka, F.; Vanderheyden, K.; De Smet, E.; Cuvelier, C.A.; Mestdagh, P.; Vandesompele, J. Straightforward and sensitive RT-qPCR based gene expression analysis of FFPE samples. Sci. Rep. 2016, 6, 21418. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Allen, L.; Altman, M.; Hlava, M.; Scott, J. Beyond authorship: Attribution, contribution, collaboration, and credit. Learn. Publ. 2015, 28, 151–155. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Patient ID | Diagnosis | FISH | mRNA Capture Sequencing | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Total Number of Uniquely Mapped Reads | Fusion Transcript (5′ Partner-3′ Partner) | Spanning Pairs a | Spanning Unique Reads b | Read Evidence Level c | Fusion Point 5′ Partner | Fusion Point 3′ Partner | |||
| P11 (cohort I) | myxoid/round cell liposarcoma | DDIT3 | 2,521,243 | FUS-DDIT3 | 4 | 6 | 3.97 | 16:31,184,396:+ (end of exon) | 12:57,517,753:− (start of exon) |
| P12 (cohort I) | Ewing sarcoma | EWSR1 | 2,837,511 | EWSR1-FLI1 | 8 | 4 | 4.23 | 22:29,288,786:+ (end of exon) | 11:128,807,180:+ (start of exon) |
| EWSR1-FLI1 | 8 | 2 | 3.52 | 22:29,291,599:+ (end of exon) | 11:128,807,180:+ (start of exon) | ||||
| P13 (cohort I) | ARMS | FOXO1 | 2,144,348 | PAX3-FOXO1 | 12 | 13 | 11.66 | 2:222,220,140:− (end of exon) | 13:40,560,860:− (start of exon) |
| P14 (cohort I) | ARMS | FOXO1 | 3,344,823 | PAX3-FOXO1 | 3 | 18 | 6.28 | 2:222,220,140:− (end of exon) | 13:40,560,860:− (start of exon) |
| P15 (cohort I) | synovial sarcoma | SS18 | 2,998,047 | SS18-SSX1 | 66 | 26 | 30.69 | 18:26,032,399:− (end of exon) | X:48,263,782:+ (start of exon) |
| P16 (cohort I) | synovial sarcoma | SS18 | 2,480,668 | SS18-SSX2 | 11 | 20 | 12.50 | 18:26,032,399:− (end of exon) | X:52,700,578:− (start of exon) |
| SS18-SSX2B | 11 | 20 | 12.50 | 18:26,032,399:− (end of exon) | X:52,757,854:+ (start of exon) | ||||
| P18 (cohort II) | ARMS (solid variant) | none | 17,582,558 | PAX3-FOXO1 | 11 | 16 | 1.54 | 2:222,220,140:− (end of exon) | 13:40,560,860:− (start of exon) |
| P25 (cohort II) | ARMS (solid variant) | none | 18,832,810 | PAX3-FOXO1 | 13 | 13 | 1.38 | 2:222,220,140:− (end of exon) | 13:40,560,860:− (start of exon) |
| P26 (cohort II) | Ewing sarcoma | none | 22,414,225 | EWSR1-ERG | 3 | 5 | 0.36 | 22:29,287,134:+ (end of exon) | 21:38,392,444:− (start of exon) |
| P27 (cohort II) | URCS; small cell osteosarcoma | none | 21,846,511 | EWSR1-NFATC2 | 19 | 12 | 1.42 | 22:29,282,557:+ (end of exon) | 20:51,516,955:− (start of exon) |
| P28 (cohort II) | URCS; small cell osteosarcoma | none | 18,512,302 | EWSR1-NFATC2 | 19 | 13 | 1.73 | 22:29,282,557:+ (end of exon) | 20:51,516,955:− (start of exon) |
| P29 (cohort II) | Ewing sarcoma | none | 21,344,451 | EWSR1-FLI1 | 4 | 5 | 0.42 | 22:29,287,134:+ (end of exon) | 11:128,805,366:+ (start of exon) |
| Patient ID | Diagnosis | mRNA Capture Sequencing | ||||||
|---|---|---|---|---|---|---|---|---|
| Total Number of Uniquely Mapped Reads | Fusion Transcript (5′ Partner-3′ Partner) | Spanning Pairs a | Spanning Unique Reads b | Read Evidence Level c | Fusion Point 5’ Partner | Fusion Point 3’ Partner | ||
| P17 | ARMS | 17,891,296 | COPS3-TOM1L2 | 11 | 9 | 1.12 | 17:17,276,035:− (end of exon) | 17:17,898,674:− (start of exon) |
| HTT-ADD1 | 7 | 5 | 0.67 | 4:3,174,799:+ (end of exon) | 4:2,904,764:+ (start of exon) | |||
| NCOA1-DTNB | 6 | 9 | 0.84 | 2:24,728,476:+ (end of exon) | 2:25,531,597:− (start of exon) | |||
| P19 | ARMS | 20,614,167 | AC138409.2-NAIP | 3 | 4 | 0.34 | 5:34,175,822:− (end of exon) | 5:71,003,857:− (start of exon) |
| NLRP2-RPL36A | 3 | 4 | 0.34 | 19:54,973,997:+ (in exon) | X:101,395,351:+ (in exon) | |||
| RPL36A-NLRP2 | 3 | 5 | 0.39 | X:101,391,808:+ (in exon) | 19:54,973,973:+ (in intron) | |||
| WWTR1-LINC01986 | 6 | 5 | 0.53 | 3:149,572,864:− (end of exon) | 3:23,968:+ (start of exon) | |||
| P20 | ARMS | 18,472,958 | AP1B1-CHEK2 | 3 | 2 | 0.27 | 22:29,349,218:− (end of exon) | 22:28,710,059:− (start of exon) |
| IGK@-BAGE2 | 54 | 17 | 3.84 | 2:89,631,593:− (end of exon) | 21:10,499,475:+ (start of exon) | |||
| PITPNC1-CACNG4 | 3 | 2 | 0.27 | 17:67,532,950:+ (end of exon) | 17:67,018,189:+ (start of exon) | |||
| PLAA-MOB3B | 8 | 3 | 0.60 | 9:26,910,338:− (end of exon) | 9:27,330,616:− (start of exon) | |||
| PTPRG-PPP4R2 | 3 | 3 | 0.32 | 3:61,562,372:+ (end of exon) | 3:72,998,077:+ (start of exon) | |||
| WDR74-ACTB | 3 | 3 | 0.32 | 11:62,837,505:− (in intron) | 7:5,527,639:− (in exon) | |||
| P22 | ARMS | 17,675,699 | GON4L-SMG5 | 2 | 5 | 0.40 | 1:155,804,949:- (end of exon) | 1:156,253,508:- (start of exon) |
| P23 | ARMS | 18,512,177 | BRD4-LEUTX | 3 | 11 | 0.76 | 19:15,254,152:− (end of exon) | 19:39,784,527:+ (start of exon) |
| P24 | ARMS | 19,914,559 | ATN1-MAML2 | 3 | 9 | 0.60 | 12:6,936,716:+ (in exon) | 11:96,092,266:− (in exon) |
| P30 | URCS | 23,211,375 | NCOA3-TBP | 3 | 3 | 0.26 | 20:47,651,098:+ (in exon) | 6:170,561,926:+ (in exon) |
| TBP-NCOA3 | 3 | 8 | 0.47 | 6:170,561,939:+ (in exon) | 20:47,651,071:+ (in exon) | |||
| P32 | URCS | 20,217,802 | MEST-RGS22 | 2 | 2 | 0.20 | 7:130,500,532:+ (end of exon) | 8:100,093,509:− (start of exon) |
| P33 | URCS | 25,928,668 | AC009021.1-RRN3 | 2 | 2 | 0.15 | 16:22,613,454:− (end of exon) | 16:15,080,096:− (start of exon) |
| Recurrently Detected Fusion (5′ Partner-3′Partner) | Disease | Patient ID | mRNA Capture Sequencing | |||||
|---|---|---|---|---|---|---|---|---|
| Total Number of Uniquely Mapped Reads | Spanning Pairs a | Spanning Unique Reads b | Read Evidence Level c | Fusion Point 5’ Partner | Fusion Point 3’ Partner | |||
| AC245595.1-IGK@ | ARMS | P19 | 27,247,781 | 10 | 2 | 0.44 | 1:144,250,225:− (end of exon) | 2:89,581,140:− (start of exon) |
| P25 | 25,800,361 | 59 | 2 | 2.36 | ||||
| ELMO1-AOAH | ARMS | P18 | 23,669,117 | 2 | 3 | 0.21 | 7:36,861,659:− (end of exon) | 7:36,674,009:− (start of exon) |
| P25 | 25,800,361 | 3 | 6 | 0.35 | ||||
| COPS4-TBC1D9 | URCS | P27 | 28,739,150 | 4 | 3 | 0.24 | 4:83,035,298:+ (end of exon) | 4:140,679,843:− (start of exon) |
| P28 | 24,863,986 | 4 | 4 | 0.32 | ||||
| PICALM-SYTL2 | URCS | P27 | 28,739,150 | 3 | 2 | 0.17 | 11:85,974,708:− (end of exon) | 11:85,758,114:− (start of exon) |
| P28 | 24,863,986 | 2 | 3 | 0.20 | ||||
| SMG6-VPS53 | URCS | P27 | 28,739,150 | 121 | 23 | 5.01 | 17:2,172,658:− (end of exon) | 17:710,613:− (start of exon) |
| P28 | 24,863,986 | 44 | 21 | 2.61 | ||||
| UBE2F-ALS2 | URCS | P27 | 28,739,150 | 6 | 13 | 0.66 | 2:237,973,225:+ (end of exon) | 2:201,718,210:− (start of exon) |
| P28 | 24,863,986 | 5 | 8 | 0.52 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decock, A.; Creytens, D.; Lefever, S.; Van der Meulen, J.; Anckaert, J.; De Ganck, A.; Deleu, J.; De Wilde, B.; Fierro, C.; Kuersten, S.; et al. mRNA Capture Sequencing and RT-qPCR for the Detection of Pathognomonic, Novel, and Secondary Fusion Transcripts in FFPE Tissue: A Sarcoma Showcase. Int. J. Mol. Sci. 2022, 23, 11007. https://doi.org/10.3390/ijms231911007
Decock A, Creytens D, Lefever S, Van der Meulen J, Anckaert J, De Ganck A, Deleu J, De Wilde B, Fierro C, Kuersten S, et al. mRNA Capture Sequencing and RT-qPCR for the Detection of Pathognomonic, Novel, and Secondary Fusion Transcripts in FFPE Tissue: A Sarcoma Showcase. International Journal of Molecular Sciences. 2022; 23(19):11007. https://doi.org/10.3390/ijms231911007
Chicago/Turabian StyleDecock, Anneleen, David Creytens, Steve Lefever, Joni Van der Meulen, Jasper Anckaert, Ariane De Ganck, Jill Deleu, Bram De Wilde, Carolina Fierro, Scott Kuersten, and et al. 2022. "mRNA Capture Sequencing and RT-qPCR for the Detection of Pathognomonic, Novel, and Secondary Fusion Transcripts in FFPE Tissue: A Sarcoma Showcase" International Journal of Molecular Sciences 23, no. 19: 11007. https://doi.org/10.3390/ijms231911007
APA StyleDecock, A., Creytens, D., Lefever, S., Van der Meulen, J., Anckaert, J., De Ganck, A., Deleu, J., De Wilde, B., Fierro, C., Kuersten, S., Luypaert, M., Rottiers, I., Schroth, G. P., Steyaert, S., Vanderheyden, K., Vanden Eynde, E., Verniers, K., Verreth, J., Van Dorpe, J., & Vandesompele, J. (2022). mRNA Capture Sequencing and RT-qPCR for the Detection of Pathognomonic, Novel, and Secondary Fusion Transcripts in FFPE Tissue: A Sarcoma Showcase. International Journal of Molecular Sciences, 23(19), 11007. https://doi.org/10.3390/ijms231911007