

Metabolic Reprogramming in Tumor Endothelial Cells

 ,
,  , and
, and 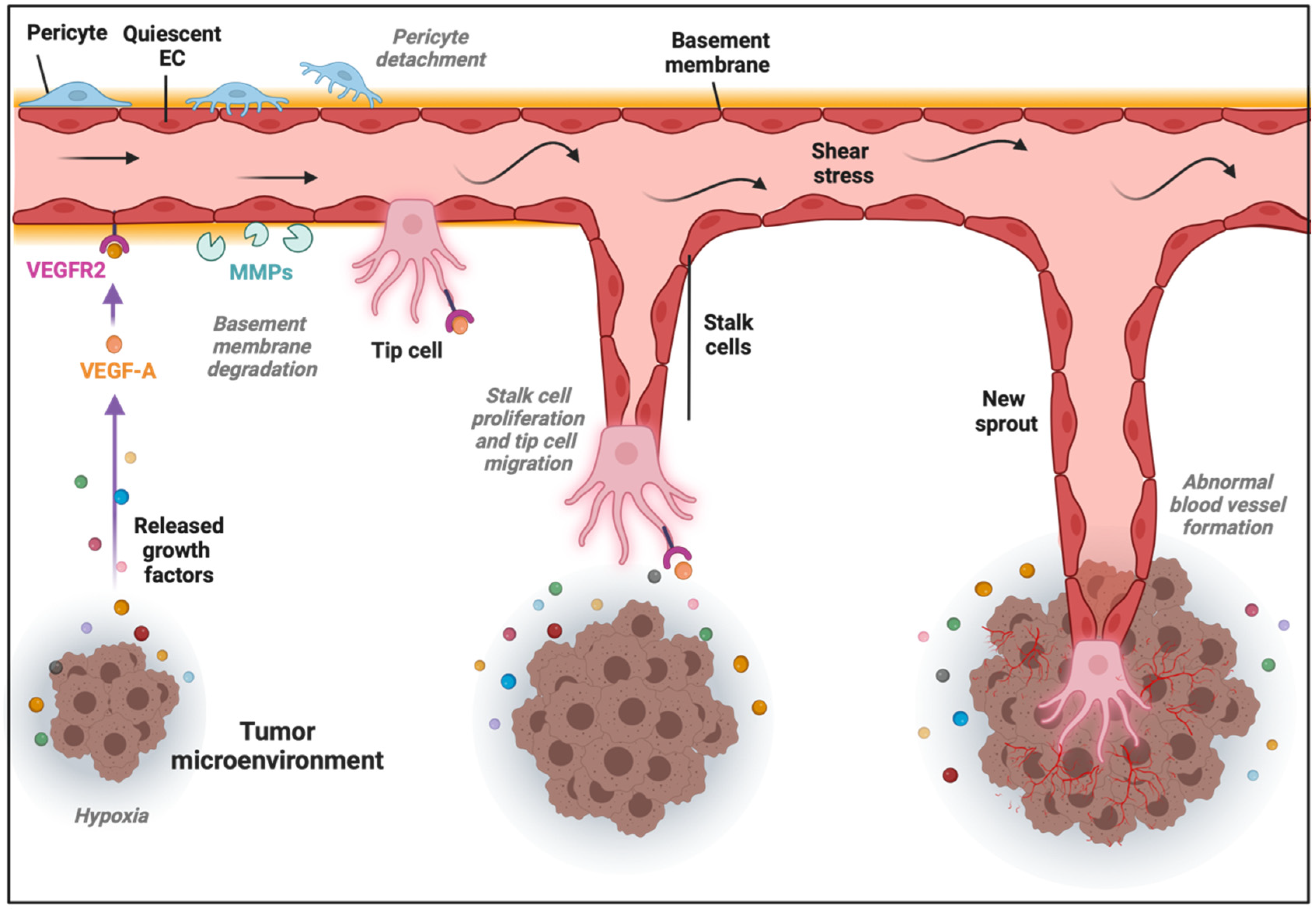{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumor Angiogenesis: A Pivotal Driver of Cancer Progression
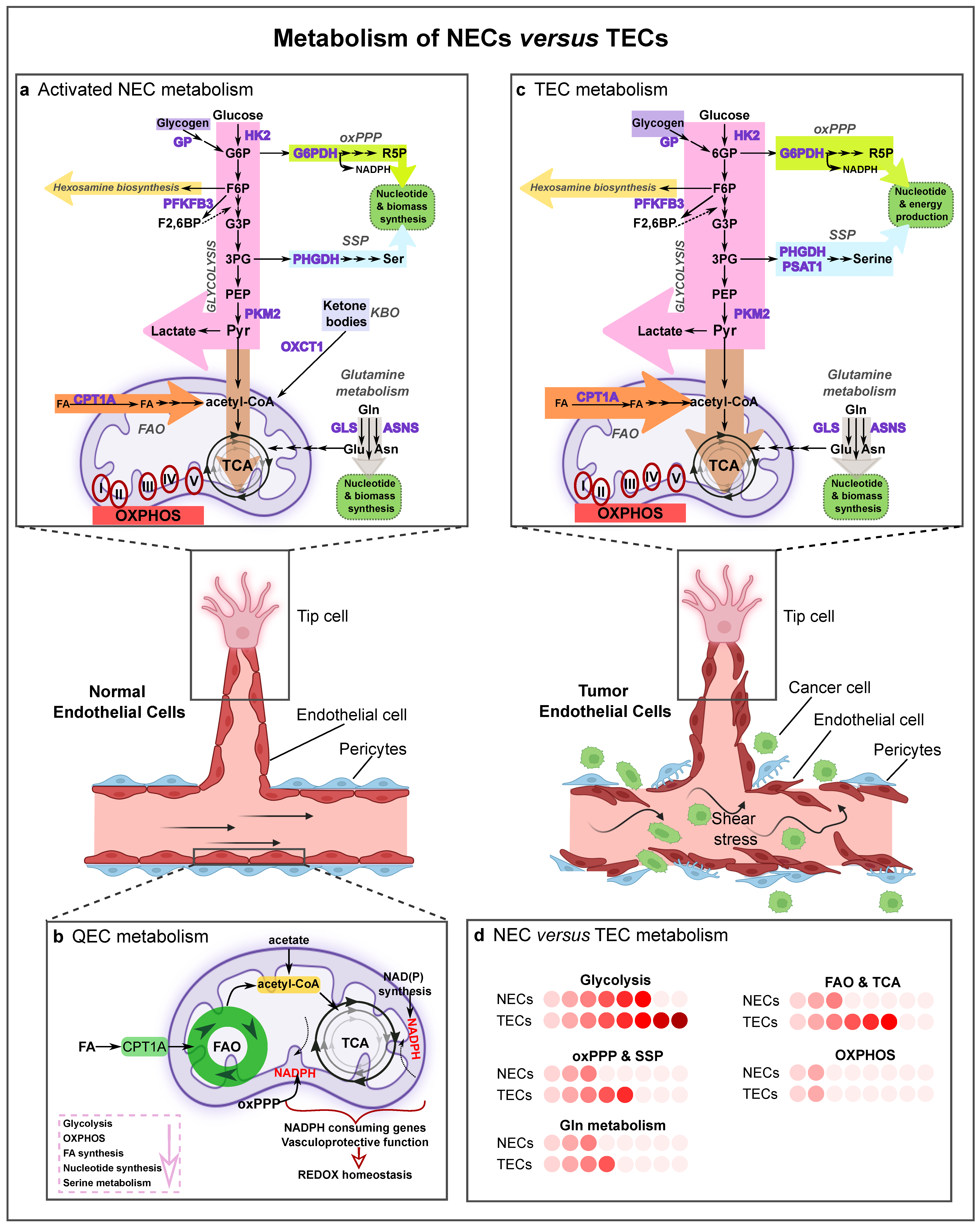
3. Metabolic Features of Normal and Tumor ECs
3.1. Normal EC (NEC) Metabolism
3.1.1. Quiescent ECs
3.1.2. Activated ECs
3.2. Tumor EC (TEC) Metabolism
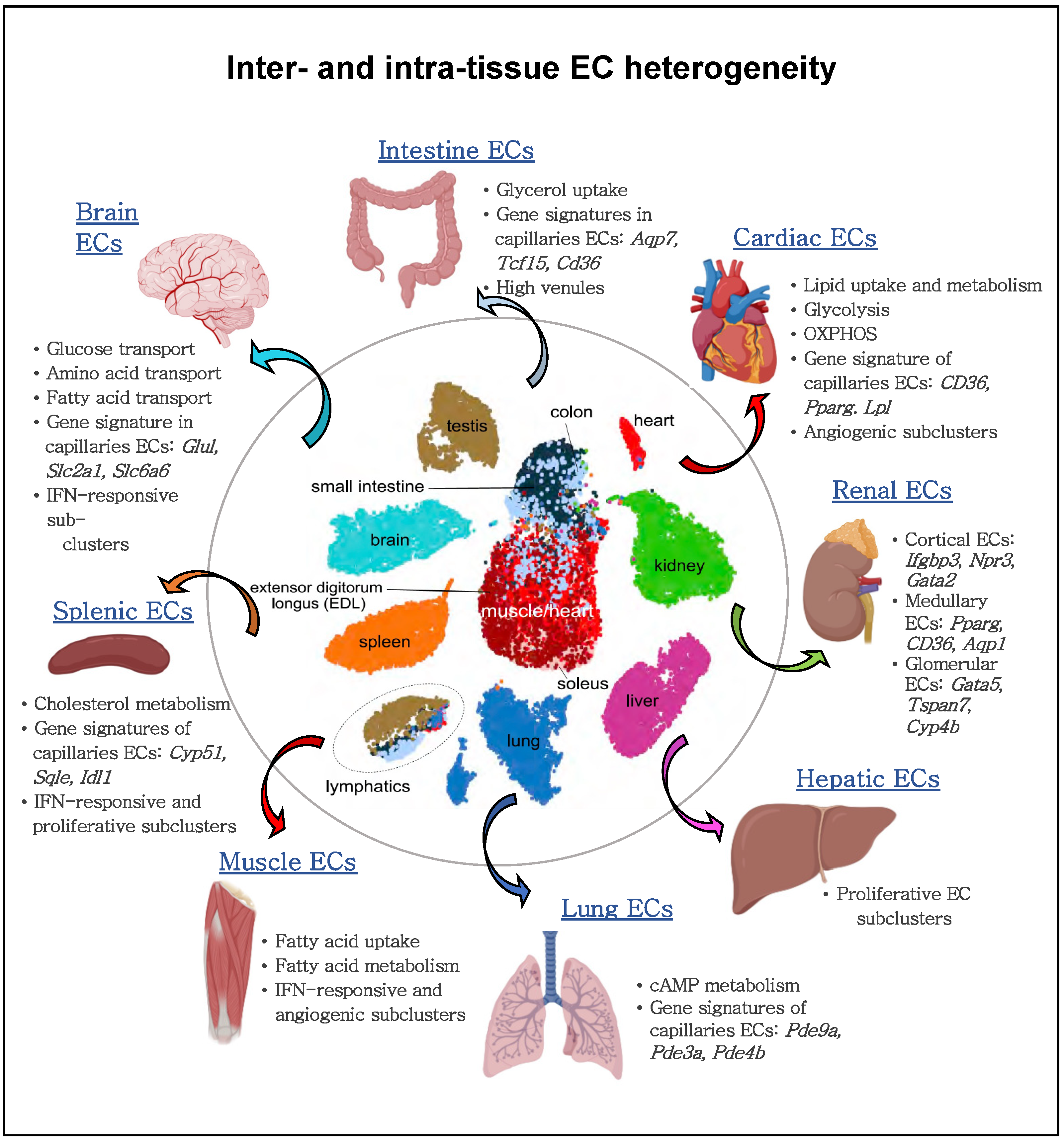
4. Metabolism of ECs at the Single-Cell Level
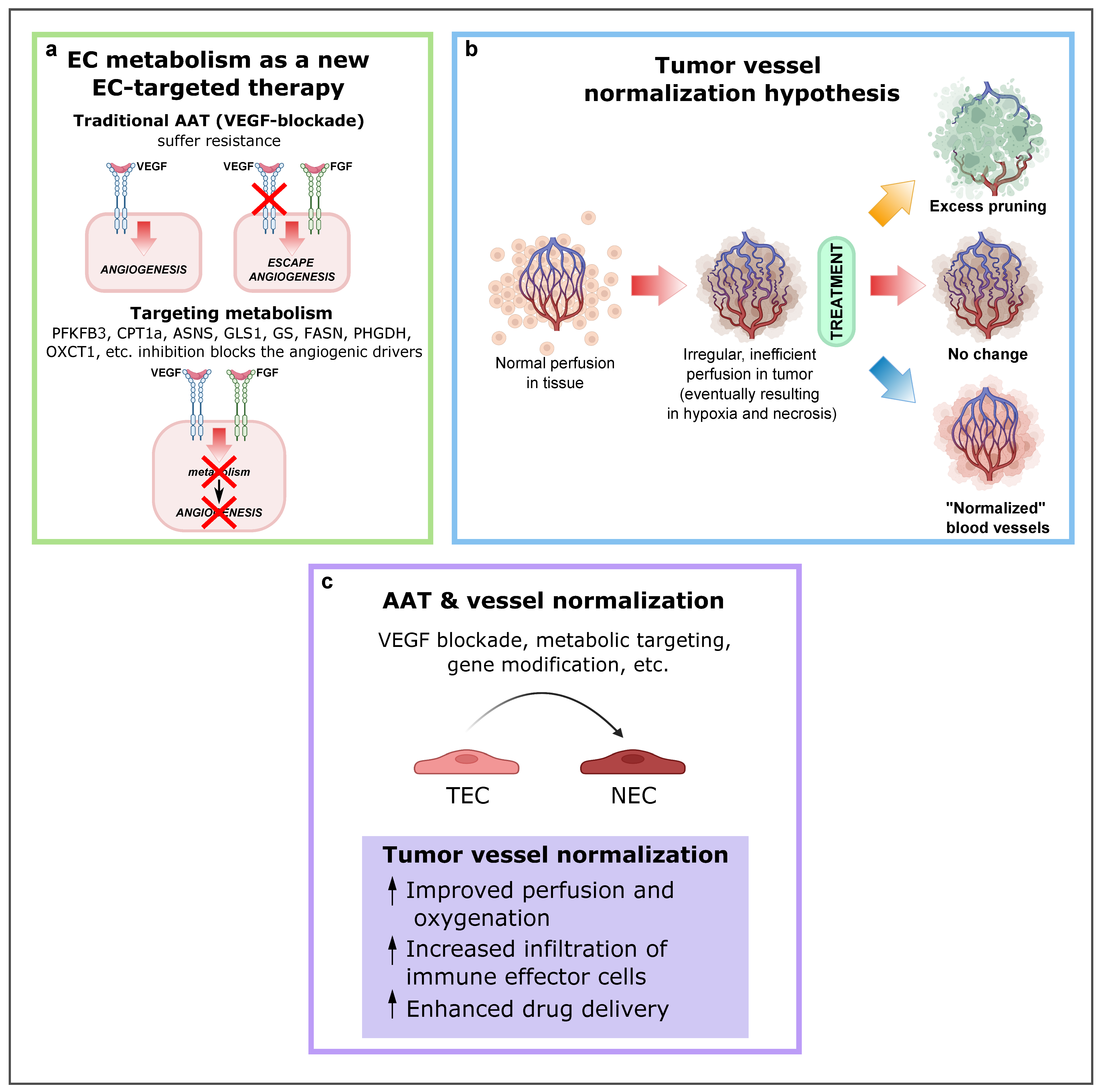
5. Modulation of Tumor Endothelial Cell Metabolism as an Innovative Therapeutic Approach
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Pircher, A.; Kalucka, J.; Carmeliet, P. Vessel pruning or healing: Endothelial metabolism as a novel target? Expert Opin. Ther. Targets 2017, 21, 239–247. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis—characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212. [Google Scholar] [CrossRef]
- Leong, S.P.; Pissas, A.; Scarato, M.; Gallon, F.; Pissas, M.H.; Amore, M.; Wu, M.; Faries, M.B.; Lund, A.W. The lymphatic system and sentinel lymph nodes: Conduit for cancer metastasis. Clin. Exp. Metastasis 2022, 39, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef]
- Zhang, L.; Li, S.; Li, L.; Chen, Z.; Yang, Y. COX2 inhibition in the endothelium induces glucose metabolism normalization and impairs tumor progression. Mol. Med. Rep. 2018, 17, 2937–2944. [Google Scholar] [CrossRef] [PubMed]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e815. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef]
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; García-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587.e513. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef]
- Mpekris, F.; Baish, J.W.; Stylianopoulos, T.; Jain, R.K. Role of vascular normalization in benefit from metronomic chemotherapy. Proc. Natl. Acad. Sci. USA 2017, 114, 1994–1999. [Google Scholar] [CrossRef]
- Martin, J.D.; Seano, G.; Jain, R.K. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu. Rev. Physiol. 2019, 81, 505–534. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Mpekris, F.; Voutouri, C.; Baish, J.W.; Duda, D.G.; Munn, L.L.; Stylianopoulos, T.; Jain, R.K. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3728–3737. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Annan, D.A.; Kikuchi, H.; Hida, Y.; Hida, K. Tumor Endothelial Heterogeneity in Cancer Progression. Cancers 2019, 11, 1511. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Siemann, D.W.; Horsman, M.R. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacol. Ther. 2015, 153, 107–124. [Google Scholar] [CrossRef]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. Embo J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Shenoy, A.K.; Lu, J. Cancer cells remodel themselves and vasculature to overcome the endothelial barrier. Cancer Lett. 2016, 380, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, H.; Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J. Immunother. Cancer 2021, 9, e002591. [Google Scholar] [CrossRef]
- Ribatti, D.; Pezzella, F. Overview on the Different Patterns of Tumor Vascularization. Cells 2021, 10, 639. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E. “Sprouting angiogenesis”, a reappraisal. Dev. Biol. 2012, 372, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; García-Caballero, M.; Carmeliet, P. Metabolic Signatures of Distinct Endothelial Phenotypes. Trends Endocrinol. Metab. 2020, 31, 580–595. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.D.; Rohlenova, K.; Luo, Y.; Carmeliet, P. The metabolic engine of endothelial cells. Nat. Metab. 2019, 1, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.C.; Pircher, A.; Geldhof, V.; de Rooij, L.; Kalucka, J.; Sokol, L.; et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020, 37, 21–36.e13. [Google Scholar] [CrossRef]
- Rohlenova, K.; Goveia, J.; García-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e814. [Google Scholar] [CrossRef]
- Saravanan, S.; Vimalraj, S.; Pavani, K.; Nikarika, R.; Sumantran, V.N. Intussusceptive angiogenesis as a key therapeutic target for cancer therapy. Life Sci. 2020, 252, 117670. [Google Scholar] [CrossRef]
- Dimova, I.; Hlushchuk, R.; Makanya, A.; Styp-Rekowska, B.; Ceausu, A.; Flueckiger, S.; Lang, S.; Semela, D.; Le Noble, F.; Chatterjee, S.; et al. Inhibition of Notch signaling induces extensive intussusceptive neo-angiogenesis by recruitment of mononuclear cells. Angiogenesis 2013, 16, 921–937. [Google Scholar] [CrossRef]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by endothelial cells—partnering up with the immune system? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Pasut, A.; Becker, L.M.; Cuypers, A.; Carmeliet, P. Endothelial cell plasticity at the single-cell level. Angiogenesis 2021, 24, 311–326. [Google Scholar] [CrossRef]
- Liu, K.; Sun, B.; Zhao, X.; Wang, X.; Li, Y.; Qiu, Z.; Gu, Q.; Dong, X.; Zhang, Y.; Wang, Y.; et al. Hypoxia induced epithelial-mesenchymal transition and vasculogenic mimicry formation by promoting Bcl-2/Twist1 cooperation. Exp. Mol. Pathol. 2015, 99, 383–391. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Teuwen, L.A.; De Rooij, L.; Cuypers, A.; Rohlenova, K.; Dumas, S.J.; García-Caballero, M.; Meta, E.; Amersfoort, J.; Taverna, F.; Becker, L.M.; et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep. 2021, 35, 109253. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef]
- Wechman, S.L.; Emdad, L.; Sarkar, D.; Das, S.K.; Fisher, P.B. Vascular mimicry: Triggers, molecular interactions and in vivo models. Adv. Cancer Res. 2020, 148, 27–67. [Google Scholar] [CrossRef]
- Seftor, R.E.; Hess, A.R.; Seftor, E.A.; Kirschmann, D.A.; Hardy, K.M.; Margaryan, N.V.; Hendrix, M.J. Tumor cell vasculogenic mimicry: From controversy to therapeutic promise. Am. J. Pathol. 2012, 181, 1115–1125. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Chao, J.T.; Chien, D.S.; Chu, Y.W. Tumor cell vascular mimicry: Novel targeting opportunity in melanoma. Pharmacol. Ther. 2016, 159, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Chen, Y.; Jiang, X.; Peng, M.; Liu, Y.; Mo, Y.; Ren, D.; Hua, Y.; Yu, B.; Zhou, Y.; et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 2021, 20, 7. [Google Scholar] [CrossRef]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197, Erratum in Nature 2015, 526, 144. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e813. [Google Scholar] [CrossRef]
- Noseda, M.; Chang, L.; McLean, G.; Grim, J.E.; Clurman, B.E.; Smith, L.L.; Karsan, A. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: Role of p21Cip1 repression. Mol. Cell Biol. 2004, 24, 8813–8822. [Google Scholar] [CrossRef] [Green Version]
- Rostama, B.; Turner, J.E.; Seavey, G.T.; Norton, C.R.; Gridley, T.; Vary, C.P.H.; Liaw, L. DLL4/Notch1 and BMP9 Interdependent Signaling Induces Human Endothelial Cell Quiescence via P27KIP1 and Thrombospondin-1. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2626–2637. [Google Scholar] [CrossRef] [PubMed]
- Dharaneeswaran, H.; Abid, M.R.; Yuan, L.; Dupuis, D.; Beeler, D.; Spokes, K.C.; Janes, L.; Sciuto, T.; Kang, P.M.; Jaminet, S.S.; et al. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ. Res. 2014, 115, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis revisited from a metabolic perspective: Role and therapeutic implications of endothelial cell metabolism. Open Biol. 2017, 7, 170219. [Google Scholar] [CrossRef]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Jang, C.; Dharaneeswaran, H.; Li, J.; Bhide, M.; Yang, S.; Li, K.; Arany, Z. Endothelial pyruvate kinase M2 maintains vascular integrity. J. Clin. Investig. 2018, 128, 4543–4556. [Google Scholar] [CrossRef]
- Helmlinger, G.; Endo, M.; Ferrara, N.; Hlatky, L.; Jain, R.K. Growth factors: Formation of endothelial cell networks. Nature 2000, 405, 139–141. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef]
- Yu, P.; Wilhelm, K.; Dubrac, A.; Tung, J.K.; Alves, T.C.; Fang, J.S.; Xie, Y.; Zhu, J.; Chen, Z.; De Smet, F.; et al. FGF-dependent metabolic control of vascular development. Nature 2017, 545, 224–228. [Google Scholar] [CrossRef]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef]
- Zecchin, A.; Kalucka, J.; Dubois, C.; Carmeliet, P. How Endothelial Cells Adapt Their Metabolism to Form Vessels in Tumors. Front. Immunol. 2017, 8, 1750. [Google Scholar] [CrossRef] [Green Version]
- Stone, O.A.; El-Brolosy, M.; Wilhelm, K.; Liu, X.; Romao, A.M.; Grillo, E.; Lai, J.K.H.; Gunther, S.; Jeratsch, S.; Kuenne, C.; et al. Loss of pyruvate kinase M2 limits growth and triggers innate immune signaling in endothelial cells. Nat. Commun. 2018, 9, 4077. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [PubMed]
- Vizán, P.; Sánchez-Tena, S.; Alcarraz-Vizán, G.; Soler, M.; Messeguer, R.; Pujol, M.D.; Lee, W.-N.P.; Cascante, M. Characterization of the metabolic changes underlying growth factor angiogenic activation: Identification of new potential therapeutic targets. Carcinogenesis 2009, 30, 946–952. [Google Scholar] [CrossRef]
- Amemiya, T. Glycogen metabolism in the capillary endothelium: Electron histochemical study of glycogen synthetase and phosphorylase in the pecten capillary of the chick. Acta Histochem. 1983, 73, 93–96. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free. Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Markowska, A.I.; Jefferies, K.C.; Panjwani, N. Galectin-3 Protein Modulates Cell Surface Expression and Activation of Vascular Endothelial Growth Factor Receptor 2 in Human Endothelial Cells*. J. Biol. Chem. 2011, 286, 29913–29921. [Google Scholar] [CrossRef]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The Notch Ligands Dll4 and Jagged1 Have Opposing Effects on Angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef]
- Lau, A.N.; Vander Heiden, M.G. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40. [Google Scholar] [CrossRef]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.E.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef]
- Vander Heiden Matthew, G.; Cantley Lewis, C.; Thompson Craig, B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Missiaen, R.; Morales-Rodriguez, F.; Eelen, G.; Carmeliet, P. Targeting endothelial metabolism for anti-angiogenesis therapy: A pharmacological perspective. Vasc. Pharmacol. 2017, 90, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; Garcia-Caballero, M.; Missiaen, R.; Huang, H.; Bruning, U.; et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 2016, 542, 49–54. [Google Scholar] [CrossRef] [PubMed]
- García-Caballero, M.; Zecchin, A.; Souffreau, J.; Truong, A.K.; Teuwen, L.A.; Vermaelen, W.; Martín-Pérez, R.; de Zeeuw, P.; Bouché, A.; Vinckier, S.; et al. Role and therapeutic potential of dietary ketone bodies in lymph vessel growth. Nat. Metab. 2019, 1, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian Folate-Mediated One-Carbon Metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef]
- Yeh, W.-L.; Lin, C.-J.; Fu, W.-M. Enhancement of Glucose Transporter Expression of Brain Endothelial Cells by Vascular Endothelial Growth Factor Derived from Glioma Exposed to Hypoxia. Mol. Pharmacol. 2008, 73, 170. [Google Scholar] [CrossRef]
- Du, W.; Ren, L.; Hamblin, M.H.; Fan, Y. Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines 2021, 9, 147. [Google Scholar] [CrossRef]
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 Plays a Critical Role in Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Seagroves, T.N.; Ryan, H.E.; Lu, H.; Wouters, B.G.; Knapp, M.; Thibault, P.; Laderoute, K.; Johnson, R.S. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol. Cell Biol. 2001, 21, 3436–3444. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.X.; Kazlauskas, A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J. Biol. Chem. 2013, 288, 21161–21172. [Google Scholar] [CrossRef] [PubMed]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef]
- Dong, L.; Krewson, E.A.; Yang, L.V. Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells. Int. J. Mol. Sci. 2017, 18, 278. [Google Scholar] [CrossRef]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef]
- Conradi, L.-C.; Brajic, A.; Cantelmo, A.R.; Bouché, A.; Kalucka, J.; Pircher, A.; Brüning, U.; Teuwen, L.-A.; Vinckier, S.; Ghesquière, B.; et al. Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis 2017, 20, 599–613. [Google Scholar] [CrossRef]
- Koziel, A.; Woyda-Ploszczyca, A.; Kicinska, A.; Jarmuszkiewicz, W. The influence of high glucose on the aerobic metabolism of endothelial EA.hy926 cells. Pflügers Arch.—Eur. J. Physiol. 2012, 464, 657–669. [Google Scholar] [CrossRef]
- Don, A.S.; Kisker, O.; Dilda, P.; Donoghue, N.; Zhao, X.; Decollogne, S.; Creighton, B.; Flynn, E.; Folkman, J.; Hogg, P.J. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell 2003, 3, 497–509. [Google Scholar] [CrossRef]
- Rohlena, J.; Dong, L.-F.; Kluckova, K.; Zobalova, R.; Goodwin, J.; Tilly, D.; Stursa, J.; Pecinova, A.; Philimonenko, A.; Hozak, P.; et al. Mitochondrially Targeted α-Tocopheryl Succinate Is Antiangiogenic: Potential Benefit Against Tumor Angiogenesis but Caution Against Wound Healing. Antioxid. Redox Signal. 2011, 15, 2923–2935. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Deng, Y.; Li, X.; Ning, Y.; Lin, X.; Guo, S.; Chen, M.; Han, M. Glutaminolysis Was Induced by TGF-β1 through PP2Ac Regulated Raf-MEK-ERK Signaling in Endothelial Cells. PLoS ONE 2016, 11, e0162658. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Carroll, P.A.; Thalhofer, A.B.; Lagunoff, M. Latent KSHV Infected Endothelial Cells Are Glutamine Addicted and Require Glutaminolysis for Survival. PLoS Pathog. 2015, 11, e1005052. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, A.P.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Filippi, I.; Saltarella, I.; Aldinucci, C.; Carraro, F.; Ria, R.; Vacca, A.; Naldini, A. Different Adaptive Responses to Hypoxia in Normal and Multiple Myeloma Endothelial Cells. Cell. Physiol. Biochem. 2018, 46, 203–212. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e720. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Li, C.; Guo, L.; Li, S.; Hua, K. Single-cell transcriptomics reveals the landscape of intra-tumoral heterogeneity and transcriptional activities of ECs in CC. Mol. Ther. Nucleic Acids 2021, 24, 682–694. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, Z.; Wang, L.; Lin, Y.; Fang, H.; Lei, J.; Cao, T.; Wang, G.; Dang, E. Single-cell transcriptome profiling reveals vascular endothelial cell heterogeneity in human skin. Theranostics 2021, 11, 6461–6476. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Singh, M.; Ferrara, N. Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat. Biotechnol. 2012, 30, 648–657. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Ruggieri, S.; Tamma, R.; Crivellato, E. Limitations of Anti-Angiogenic Treatment of Tumors. Transl. Oncol. 2019, 12, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- Ashrafian, H.; Horowitz, J.D.; Frenneaux, M.P. Perhexiline. Cardiovasc. Drug Rev. 2007, 25, 76–97. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, M.N.; Butterworth, E.A.; Kilberg, M.S. Asparagine synthetase: Regulation by cell stress and involvement in tumor biology. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E789–E799. [Google Scholar] [CrossRef] [PubMed]
- Newburg, D.S.; Frankel, D.L.; Fillios, L.C. An asparagine requirement in young rats fed the dietary combinations of aspartic acid, glutamine, and glutamic acid. J. Nutr. 1975, 105, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Averous, J.; Bruhat, A.; Carraro, V.; Mordier, S.; Fafournoux, P. Amino acids as regulators of gene expression: Molecular mechanisms. Biochem. Biophys. Res. Commun. 2004, 313, 447–452. [Google Scholar] [CrossRef]
- Scioscia, K.A.; Snyderman, C.H.; Wagner, R. Altered serum amino acid profiles in head and neck cancer. Nutr. Cancer 1998, 30, 144–147. [Google Scholar] [CrossRef]
- Ahlman, B.; Andersson, K.; Leijonmarck, C.E.; Ljungqvist, O.; Hedenborg, L.; Wernerman, J. Short-term starvation alters the free amino acid content of the human intestinal mucosa. Clin. Sci. 1994, 86, 653–662. [Google Scholar] [CrossRef]
- Bastos, D.C.; Paupert, J.; Maillard, C.; Seguin, F.; Carvalho, M.A.; Agostini, M.; Coletta, R.D.; Noël, A.; Graner, E. Effects of fatty acid synthase inhibitors on lymphatic vessels: An in vitro and in vivo study in a melanoma model. Lab. Investig. 2017, 97, 194–206. [Google Scholar] [CrossRef]
- Sorensen, A.G.; Emblem, K.E.; Polaskova, P.; Jennings, D.; Kim, H.; Ancukiewicz, M.; Wang, M.; Wen, P.Y.; Ivy, P.; Batchelor, T.T.; et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012, 72, 402–407, Correction in Cancer Res. 2012, 72, 1316. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Caballero, M.; Sokol, L.; Cuypers, A.; Carmeliet, P. Metabolic Reprogramming in Tumor Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 11052. https://doi.org/10.3390/ijms231911052
García-Caballero M, Sokol L, Cuypers A, Carmeliet P. Metabolic Reprogramming in Tumor Endothelial Cells. International Journal of Molecular Sciences. 2022; 23(19):11052. https://doi.org/10.3390/ijms231911052
Chicago/Turabian StyleGarcía-Caballero, Melissa, Liliana Sokol, Anne Cuypers, and Peter Carmeliet. 2022. "Metabolic Reprogramming in Tumor Endothelial Cells" International Journal of Molecular Sciences 23, no. 19: 11052. https://doi.org/10.3390/ijms231911052
APA StyleGarcía-Caballero, M., Sokol, L., Cuypers, A., & Carmeliet, P. (2022). Metabolic Reprogramming in Tumor Endothelial Cells. International Journal of Molecular Sciences, 23(19), 11052. https://doi.org/10.3390/ijms231911052