

Metagenomic Sequencing for the Diagnosis of Plasmodium spp. with Different Levels of Parasitemia in EDTA Blood of Malaria Patients—A Proof-of-Principle Assessment
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
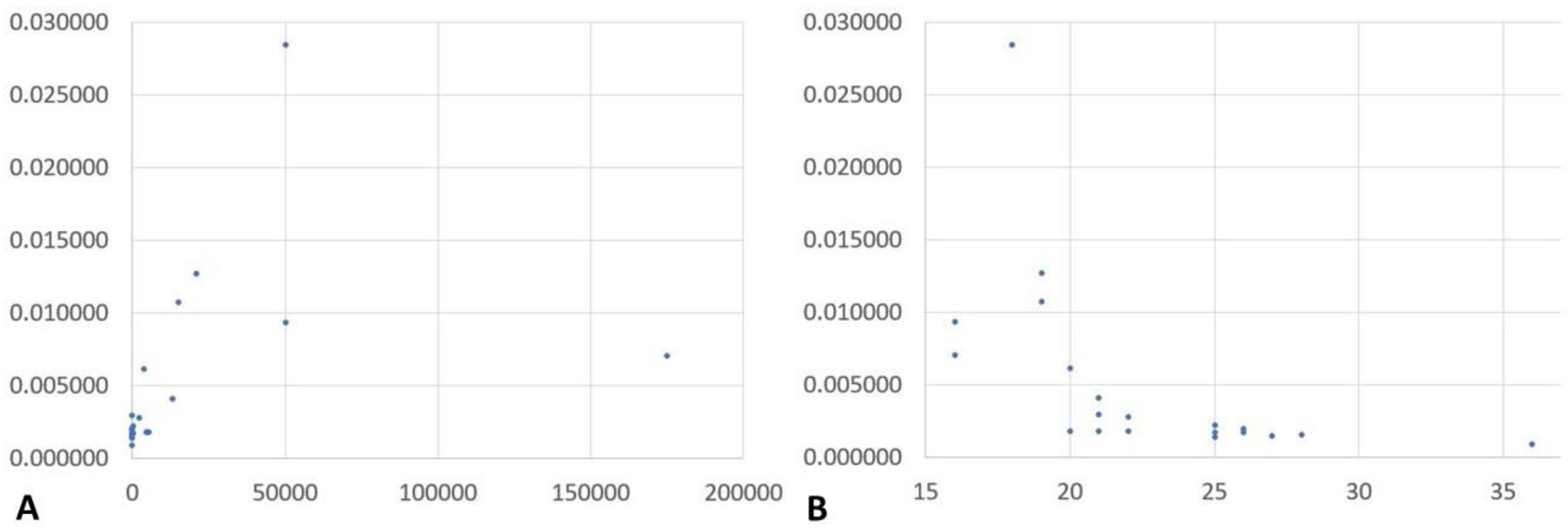
2.1. NGS Results and Correlation of Reads Assigned to Plasmodium spp. with Microscopically Assessed Parasitemia and Plasmodium Genus-Specific Real-Time PCR-Based Semi-Quantification
2.2. Matching of the Diagnostic Results on Species Level Based on the Diagnostic Reference Approach and Bioinformatic Assessments Based on Kraken, Bracken and Pavian
3. Discussion
4. Materials and Methods
4.1. Study Design and Sample Materials
4.2. Metagenomic Next Generation Sequencing
4.3. Bioinformatic Sequence Analysis and Statistics
4.4. Ethical Clearance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leder, K.; Torresi, J.; Brownstein, J.S.; Wilson, M.E.; Keystone, J.S.; Barnett, E.; Schwartz, E.; Schlagenhauf, P.; Wilder-Smith, A.; Castelli, F.; et al. GeoSentinel Surveillance Network. Travel-associated illness trends and clusters, 2000–2010. Emerg. Infect. Dis. 2013, 19, 1049–1073. [Google Scholar] [CrossRef]
- Leder, K.; Torresi, J.; Libman, M.D.; Cramer, J.P.; Castelli, F.; Schlagenhauf, P.; Wilder-Smith, A.; Wilson, M.E.; Keystone, J.S.; Schwartz, E.; et al. GeoSentinel Surveillance Network. GeoSentinel surveillance of illness in returned travelers, 2007–2011. Ann. Intern. Med. 2013, 158, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Giemsa, G. Eine Vereinfachung und Vervollkommnung meiner Methylenblau-Eosin-Färbemethode zur Erzielung der Romanowsky-Nocht’schen Chromatinfärbung. Cent. Bakteriol. 1904, 32, 307–313. [Google Scholar]
- Altangerel, E.; Frickmann, H. Meta-analysis of the diagnostic performance characteristics of three commercial and one in-house nucleic acid amplification tests for malaria screening. J. Lab. Med. 2020, 44, 47–53. [Google Scholar] [CrossRef]
- Mischlinger, J.; Pitzinger, P.; Veletzky, L.; Groger, M.; Zoleko-Manego, R.; Adegnika, A.A.; Agnandji, S.T.; Lell, B.; Kremsner, P.G.; Tannich, E.; et al. Use of Capillary Blood Samples Leads to Higher Parasitemia Estimates and Higher Diagnostic Sensitivity of Microscopic and Molecular Diagnostics of Malaria Than Venous Blood Samples. J. Infect. Dis. 2018, 218, 1296–1305. [Google Scholar] [CrossRef]
- Shokoples, S.E.; Ndao, M.; Kowalewska-Grochowska, K.; Yanow, S.K. Multiplexed real-time PCR assay for discrimination of Plasmodium species with improved sensitivity for mixed infections. J. Clin. Microbiol. 2009, 47, 975–980. [Google Scholar] [CrossRef]
- Grossman, T.; Schwartz, E.; Vainer, J.; Agmon, V.; Glazer, Y.; Goldmann, D.; Marva, E. Contribution of real-time PCR to Plasmodium species identification and to clinical decisions: A nationwide study in a non-endemic setting. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 671–675. [Google Scholar] [CrossRef]
- Frickmann, H.; Wegner, C.; Ruben, S.; Behrens, C.; Kollenda, H.; Hinz, R.; Rojak, S.; Schwarz, N.G.; Hagen, R.M.; Tannich, E. Evaluation of the multiplex real-time PCR assays RealStar malaria S&T PCR kit 1.0 and FTD malaria differentiation for the differentiation of Plasmodium species in clinical samples. Travel Med. Infect. Dis. 2019, 31, 101442. [Google Scholar]
- Kotepui, M.; Masangkay, F.R.; Kotepui, K.U.; De Jesus Milanez, G. Misidentification of Plasmodium ovale as Plasmodium vivax malaria by a microscopic method: A meta-analysis of confirmed P. ovale cases. Sci. Rep. 2020, 10, 21807. [Google Scholar] [CrossRef]
- Calderaro, A.; Piccolo, G.; Perandin, F.; Gorrini, C.; Peruzzi, S.; Zuelli, C.; Ricci, L.; Manca, N.; Dettori, G.; Chezzi, C.; et al. Genetic polymorphisms influence Plasmodium ovale PCR detection accuracy. J. Clin. Microbiol. 2007, 45, 1624–1627. [Google Scholar] [CrossRef]
- Calderaro, A.; Piccolo, G.; Gorrini, C.; Montecchini, S.; Rossi, S.; Medici, M.C.; Chezzi, C.; Snounou, G. A new real-time PCR for the detection of Plasmodium ovale wallikeri. PLoS ONE 2012, 7, e48033. [Google Scholar] [CrossRef] [PubMed]
- Bauffe, F.; Desplans, J.; Fraisier, C.; Parzy, D. Real-time PCR assay for discrimination of Plasmodium ovale curtisi and Plasmodium ovale wallikeri in the Ivory Coast and in the Comoros Islands. Malar. J. 2012, 11, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frickmann, H.; Wegner, C.; Ruben, S.; Loderstädt, U.; Tannich, E. A comparison of two PCR protocols for the differentiation of Plasmodium ovale species and implications for clinical management in travellers returning to Germany: A 10-year cross-sectional study. Malar. J. 2019, 18, 272. [Google Scholar] [CrossRef]
- Bourgeois, N.; Boutet, A.; Bousquet, P.J.; Basset, D.; Douard-Enault, C.; Charachon, S.; Lachaud, L. Comparison of three real-time PCR methods with blood smears and rapid diagnostic test in Plasmodium sp. infection. Clin. Microbiol. Infect. 2010, 16, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Dormond, L.; Jaton-Ogay, K.; de Vallière, S.; Genton, B.; Bille, J.; Greub, G. Multiplex real-time PCR for the diagnosis of malaria: Correlation with microscopy. Clin. Microbiol. Infect. 2011, 17, 469–475. [Google Scholar] [CrossRef]
- Picot, S.; Cucherat, M.; Bienvenu, A.L. Systematic review and meta-analysis of diagnostic accuracy of loop-mediated isothermal amplification (LAMP) methods compared with microscopy, polymerase chain reaction and rapid diagnostic tests for malaria diagnosis. Int. J. Infect. Dis. 2020, 98, 408–419. [Google Scholar] [CrossRef]
- Deen, J.; Mukaka, M.; von Seidlein, L. What is the yield of malaria reactive case detection in the Greater Mekong Sub-region? A review of published data and meta-analysis. Malar. J. 2021, 20, 131. [Google Scholar] [CrossRef]
- Kamaliddin, C.; Sutherland, C.J.; Houze, S.; Cottrell, G.; Briand, V.; Mogollon, D.C.; Pillai, D.R. The role of ultra-sensitive molecular methods for detecting malaria—The broader perspective. Clin. Infect. Dis. 2021, 73, e1387–e1390. [Google Scholar] [CrossRef]
- Zainabadi, K. Ultrasensitive Diagnostics for Low-Density Asymptomatic Plasmodium falciparum Infections in Low-Transmission Settings. J. Clin. Microbiol. 2021, 59, e01508-20. [Google Scholar] [CrossRef]
- Whittaker, C.; Slater, H.; Nash, R.; Bousema, T.; Drakeley, C.; Ghani, A.C.; Okell, L.C. Global patterns of submicroscopic Plasmodium falciparum malaria infection: Insights from a systematic review and meta-analysis of population surveys. Lancet Microbe 2021, 2, e366–e374. [Google Scholar] [CrossRef]
- Mbanefo, A.; Kumar, N. Evaluation of Malaria Diagnostic Methods as a Key for Successful Control and Elimination Programs. Trop. Med. Infect. Dis. 2020, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Prusty, D.; Gupta, N.; Upadhyay, A.; Dar, A.; Naik, B.; Kumar, N.; Prajapati, V.K. Asymptomatic malaria infection prevailing risks for human health and malaria elimination. Infect. Genet. Evol. 2021, 93, 104987. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Podbielski, A.; Meyer, T.; Zautner, A.E.; Loderstädt, U.; Schwarz, N.G.; Krüger, A.; Cadar, D.; Frickmann, H. On detection thresholds-a review on diagnostic approaches in the infectious disease laboratory and the interpretation of their results. Acta Trop. 2020, 205, 105377. [Google Scholar] [CrossRef] [PubMed]
- Loderstädt, U.; Hagen, R.M.; Hahn, A.; Frickmann, H. New Developments in PCR-Based Diagnostics for Bacterial Pathogens Causing Gastrointestinal Infections-A Narrative Mini-Review on Challenges in the Tropics. Trop. Med. Infect. Dis. 2021, 6, 96. [Google Scholar] [CrossRef]
- Luciani, F.; Bull, R.A.; Lloyd, A.R. Next generation deep sequencing and vaccine design: Today and tomorrow. Trends Biotechnol. 2012, 30, 443–452. [Google Scholar] [CrossRef]
- Bhatt, A.S.; Freeman, S.S.; Herrera, A.F.; Pedamallu, C.S.; Gevers, D.; Duke, F.; Jung, J.; Michaud, M.; Walker, B.J.; Young, S.; et al. Sequence-based discovery of Bradyrhizobium enterica in cord colitis syndrome. N. Engl. J. Med. 2013, 369, 517–528. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Giombini, E.; Rozera, G. Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013, 19, 15–22. [Google Scholar] [CrossRef]
- Fischer, N.; Rohde, H.; Indenbirken, D.; Günther, T.; Reumann, K.; Lütgehetmann, M.; Meyer, T.; Kluge, S.; Aepfelbacher, M.; Alawi, M.; et al. Rapid metagenomic diagnostics for suspected outbreak of severe pneumonia. Emerg. Infect. Dis. 2014, 20, 1072–1075. [Google Scholar] [CrossRef]
- Fischer, N.; Indenbirken, D.; Meyer, T.; Lütgehetmann, M.; Lellek, H.; Spohn, M.; Aepfelbacher, M.; Alawi, M.; Grundhoff, A. Evaluation of Unbiased Next-Generation Sequencing of RNA (RNA-seq) as a Diagnostic Method in Influenza Virus-Positive Respiratory Samples. J. Clin. Microbiol. 2015, 53, 2238–2250. [Google Scholar] [CrossRef]
- Boonham, N.; Kreuze, J.; Winter, S.; van der Vlugt, R.; Bergervoet, J.; Tomlinson, J.; Mumford, R. Methods in virus diagnostics: From ELISA to next generation sequencing. Virus Res. 2014, 186, 20–31. [Google Scholar] [CrossRef]
- Quinones-Mateu, M.E.; Avila, S.; Reyes-Teran, G.; Martinez, M.A. Deep sequencing: Becoming a critical tool in clinical virology. J. Clin. Virol. 2014, 61, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Di Resta, C.; Galbiati, S.; Carrera, P.; Ferrari, M. Next-generation sequencing approach for the diagnosis of human diseases: Open challenges and new opportunities. Ejifcc 2018, 29, 4–14. [Google Scholar] [PubMed]
- Xu, Y.; Lewandowski, K.; Downs, L.O.; Kavanagh, J.; Hender, T.; Lumley, S.; Jeffery, K.; Foster, D.; Sanderson, N.D.; Vaughan, A.; et al. Nanopore metagenomic sequencing of influenza virus directly from respiratory samples: Diagnosis, drug resistance and nosocomial transmission, United Kingdom, 2018/19 influenza season. Eurosurveillance 2021, 26, 2000004. [Google Scholar] [CrossRef] [PubMed]
- Khairat, R.; Ball, M.; Chang, C.C.; Bianucci, R.; Nerlich, A.G.; Trautmann, M.; Ismail, S.; Shanab, G.M.; Karim, A.M.; Gad, Y.Z.; et al. First insights into the metagenome of Egyptian mummies using next-generation sequencing. J. Appl. Genet. 2013, 54, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Jerome, H.; Taylor, C.; Sreenu, V.B.; Klymenko, T.; Filipe, A.D.S.; Jackson, C.; Davis, C.; Ashraf, S.; Wilson-Davies, E.; Jesudason, N.; et al. Metagenomic next-generation sequencing aids the diagnosis of viral infections in febrile returning travellers. J. Infect. 2019, 79, 383–388. [Google Scholar] [CrossRef]
- Ramesh, A.; Nakielny, S.; Hsu, J.; Kyohere, M.; Byaruhanga, O.; de Bourcy, C.; Egger, R.; Dimitrov, B.; Juan, Y.F.; Sheu, J.; et al. Metagenomic next-generation sequencing of samples from pediatric febrile illness in Tororo, Uganda. PLoS ONE 2019, 14, e0218318. [Google Scholar] [CrossRef]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.F.; King, R.; et al. IDseq-An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience 2020, 9, giaa111. [Google Scholar] [CrossRef]
- Sherry, N.L.; Porter, J.L.; Seemann, T.; Watkins, A.; Stinear, T.P.; Howden, B.P. Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J. Clin. Microbiol. 2013, 51, 1396–1401. [Google Scholar] [CrossRef]
- Frickmann, H.; Künne, C.; Hagen, R.M.; Podbielski, A.; Normann, J.; Poppert, S.; Looso, M.; Kreikemeyer, B. Next-generation sequencing for hypothesis-free genomic detection of invasive tropical infections in poly-microbially contaminated, formalin-fixed, paraffin-embedded tissue samples—A proof-of-principle assessment. BMC Microbiol. 2019, 19, 75. [Google Scholar] [CrossRef]
- Lu, J.; Salzberg, S.L. Removing contaminants from databases of draft genomes. PLOS Comput. Biol. 2018, 14, e1006277. [Google Scholar] [CrossRef]
- Wright, R.J.; Comeau, A.M.; Langille, M.G.I. From defaults to databases: Parameter and database choice dramatically impact the performance of metagenomic taxonomic classification tools. BioRxiv 2022. [Google Scholar] [CrossRef]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Cornet, L.; Baurain, D. Contamination detection in genomic data: More is not enough. Genome Biol. 2022, 23, 60. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 2020, 36, 1303–1304. [Google Scholar] [CrossRef]
- Li, Z.; Breitwieser, F.P.; Lu, J.; Jun, A.S.; Asnaghi, L.; Salzberg, S.L.; Eberhart, C.G. Identifying Corneal Infections in Formalin-Fixed Specimens Using Next Generation Sequencing. Investig. Opthalmology Vis. Sci. 2018, 59, 280. [Google Scholar] [CrossRef]
- Frickmann, H.; Hinz, R.; Rojak, S.; Bonow, I.; Ruben, S.; Wegner, C.; Zielke, I.; Hagen, R.M.; Tannich, E. Evaluation of automated loop-mediated amplification (LAMP) for routine malaria detection in blood samples of German travelers—A cross sectional study. Travel Med. Infect. Dis. 2018, 24, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Mangold, K.A.; Manson, R.U.; Koay, E.S.; Stephens, L.; Regner, M.; Thomson, R.B., Jr.; Peterson, L.R.; Kaul, K.L. Real-time PCR for detection and identification of Plasmodium spp. J. Clin. Microbiol. 2005, 43, 2435–2440. [Google Scholar] [CrossRef]
- Hagen, R.M.; Hinz, R.; Tannich, E.; Frickmann, H. Comparison of two real-time PCR assays for the detection of malaria parasites from hemolytic blood samples—Short communication. Eur. J. Microbiol. Immunol. 2015, 5, 159–163. [Google Scholar] [CrossRef]
- Frickmann, H.; Lunardon, L.M.; Hahn, A.; Loderstädt, U.; Lindner, A.K.; Becker, S.L.; Mockenhaupt, F.P.; Weber, C.; Tannich, E. Evaluation of a duplex real-time PCR in human serum for simultaneous detection and differentiation of Schistosoma mansoni and Schistosoma haematobium infections—Cross-sectional study. Travel. Med. Infect. Dis. 2021, 41, 102035. [Google Scholar] [CrossRef]
- Bossuyt, P.M.; Reitsma, J.B.; Bruns, D.E.; Gatsonis, C.A.; Glasziou, P.P.; Irwig, L.; Lijmer, J.G.; Moher, D.; Rennie, D.; De Vet, H.C.W.; et al. STARD 2015: An updated list of essential items for reporting diagnostic accuracy studies. BMJ 2015, 351, h5527. [Google Scholar] [CrossRef] [PubMed]
- Frickmann, H.; Hinz, R.; Hagen, R.M. Comparison of an automated nucleic acid extraction system with the column-based procedure. Eur. J. Microbiol. Immunol. 2015, 5, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, K.L.; Contreras-Moreira, B.; de Silva, N.; Maslen, G.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Barba, M.; Bolser, D.M.; Cambell, L.; et al. Ensembl Genomes 2020—Enabling non-vertebrate genomic research. Nucleic Acids Res. 2020, 48, D689–D695. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B. Pyfasta (Version 0.5.2). Available online: https://github.com/brentp/pyfasta (accessed on 23 August 2022).
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Sample Code | Parasitemia with Plasmodium spp. as Assessed by Microscopy | Cycle Threshold Value in Genus Species PCR for Plasmodium spp. | Number of Total Reads | Number of Reads Assigned to Homo sapiens, in Brackets: Percentage of Total Reads | Number of Reads Assigned to Plasmodium spp., in Brackets: Percentage of Total Reads |
|---|---|---|---|---|---|
| N.C. | 0/µL | n.a. | 25,002,603 | 24,515,911 (98.05%) | 53,342 (0.21%) |
| D016 | 50.000/µL | 16 | 13,682,913 | 13,303,308 (97.23%) | 129,035 (0.94%) |
| D169 | <50/µL | 28 | 22,763,368 | 22,249,906 (97.74%) | 37,024 (0.16%) |
| D170 | 272/µL | 25 | 23,842,401 | 23,364,531 (98.00%) | 52,209 (0.22%) |
| D178 | 175.000/µL | 16 | 27,137,956 | 26,494,085 (97.63%) | 192,375 (0.71%) |
| D020 | 50/µL | 26 | 20,402,946 | 20,018,621 (98.12%) | 40,501 (0.20%) |
| D216 | 50.000/µL | 18 | 16,695,865 | 16,007,009 (95.87%) | 475,761 (2.85%) |
| D225 | 14.920/µL | 19 | 26,345,466 | 25,669,538 (97.43%) | 283,245 (1.08%) |
| D234 | 4.000/µL | 20 | 17,687,032 | 17,412,071 (98.45%) | 110,231 (0.62%) |
| D270 | <50/µL | 27 | 17,725,864 | 17,532,908 (98.91%) | 26,938 (0.15%) |
| D272 | 104/µL | 26 | 24,323,327 | 23,844,801 (98.03%) | 40,256 (0.17%) |
| D282 | 122/µL | 25 | 15,819,159 | 15,634,285 (98.83%) | 26,959 (0.17%) |
| D293 | 5.000/µL | 20 | 19,060,562 | 18,855,573 (98.92%) | 34,745 (0.18%) |
| D302 | 50/µL | 36 | 20,613,039 | 20,353,551 (98.74%) | 19,257 (0.09%) |
| D417 | 13.000/µL | 21 | 22,729,535 | 22,409,043 (98.59%) | 93,498 (0.41%) |
| D465 | <50/µL | negative ^ | 23,225,621 | 22,888,629 (98.55%) | 38,203 (0.16%) |
| D503 | 50/µL | 21 | 25,935,179 | 25,647,132 (98.89%) | 78,501 (0.30%) |
| D558 | 2.400/µL | 22 | 24,659,561 | 24,346,841 (98.73%) | 69,882 (0.28%) |
| D567 * | 4.600/µL | 22 | 28,609,738 | 27,803,374 (97.18%) | 50,884 (0.18%) |
| D570 | 5.240/µL | 21 | 18,913,510 | 18,713,593 (98.94%) | 34,637 (0.18%) |
| D583 * | 50/µL | 25 | 31,978,731 | 31,467,597 (98.40%) | 43,995 (0.14%) |
| D747 ° | 20.920/µL | 19 | 23,097,171 | 22,652,992 (98.08%) | 293,161 (1.27%) |
| Sample Code | Species According to the Reference Diagnostic Approach | Plasmodium Species Applying the Kraken Approach (Number of Assigned Reads) | Plasmodium Species Applying the Bracken Approach (Number of Assigned Reads) | Plasmodium Species Applying the Pavian Approach (Calculated Z-Score) |
|---|---|---|---|---|
| N.C. | Schistosoma mansoni | Plasmodium ovale wallikeri (46,154) | Plasmodium ovale complex (48,062) | Plasmodium gallinaceum (3.4) |
| D016 | Plasmodium falciparum | P. falciparum (65,171) | P. falciparum (103,363) | P. falciparum (21,980.0) |
| D169 | P. falciparum | P. ovale wallikeri (29,240) | P. ovale complex (30,597) | P. falciparum (10.5) |
| D170 | Plasmodium vivax | P. ovale wallikeri (36,296) | P. ovale complex (36,872) | P. vivax (4.3) |
| D178 | P. falciparum | P. falciparum (105,104) | P. falciparum (168,368) | P. falciparum (35,440.0) |
| D020 | Plasmodium falciparum | P. ovale wallikeri (30,068) | P. ovale complex (31,501) | P. vivax (52.3) |
| D216 | P. vivax | P. vixax (380,816) | P. vivax (457,652) | P. vivax (374.8) |
| D225 | P. vivax | P. vivax (222,565) | P. vivax (259,235) | P. vivax (218.4) |
| D234 | P. vivax | P. vivax (82,354) | P. vivax (98,480) | P. vivax (79.9) |
| D270 | Plasmodium malariae | P. ovale wallikeri (21,138) | P. ovale complex (21,682) | Plasmodium relictum (1.0) |
| D272 | P. malariae | P. ovale wallikeri (32,768) | P. ovale complex (34,566) | P. falciparum (3.7) |
| D282 | P. malariae | P. ovale wallikeri (20,891) | P. ovale complex (21,482) | P. malariae (0.3) |
| D293 | P. ovale curtisi | P. ovale curtisi (18,404) | P. ovale complex (31,708) | P. ovale complex (128.4) |
| D302 | P. ovale complex | P. ovale wallikeri (13,743) | P. ovale complex (13,820) | P. vivax (2.0) |
| D417 | Plasmodium ovale curtisi | P. ovale curtisi (18,404) | P. ovale complex (90,625) | Plasmodium ovale complex (404.5) |
| D465 | P. malariae | P. ovale wallikeri (28,460) | P. ovale complex (30,082) | Plasmodium spp. (1.0) |
| D503 | P. ovale curtisi | P. ovale curtisi (55,869) | P. ovale complex (77,118) | P. ovale complex (390.5) |
| D558 | P. vivax | P. vivax (35,625) | P. vivax (43,473) | P. vivax (33.7) |
| D567 * | P. falciparum & P. vivax | P. ovale wallikeri (29,901) | P. ovale complex (31,972) | P. falciparum (390.5) |
| D570 | P. falciparum & P. vivax | P. ovale wallikeri (23,969) | P. ovale complex (24,521) | P. falciparum (2241.0) |
| D583 * | P. falciparum & P. vivax | P. ovale wallikeri (33,528) | P. ovale complex (35,859) | P. falciparum (136.2) |
| D747 ° | P. ovale curtisi | P. ovale curtisi (263,024) | P. ovale complex (291,205) | P. ovale complex (1648.0) |
| Microscopy | Traditional Molecular Diagnostic Approaches (e.g., Real-Time PCR, Loop-Mediated Isothermal Amplification) | Diagnostic Application of the Described Next Generation Sequencing Approach | |
|---|---|---|---|
| Time required for the diagnostic workflow | About 1 h | 1 to few hours | 2–3 days |
| Hands-on-time | About 1 h | Few minutes to 1 h | Several hours |
| Reagent costs per sample | Less than 1 US dollar | Less than 100 US dollars | More than 1000 US dollars |
|
| ↓ |
|
| ↓ |
|
| ↓ |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frickmann, H.; Weinreich, F.; Loderstädt, U.; Poppert, S.; Tannich, E.; Bull, J.; Kreikemeyer, B.; Barrantes, I. Metagenomic Sequencing for the Diagnosis of Plasmodium spp. with Different Levels of Parasitemia in EDTA Blood of Malaria Patients—A Proof-of-Principle Assessment. Int. J. Mol. Sci. 2022, 23, 11150. https://doi.org/10.3390/ijms231911150
Frickmann H, Weinreich F, Loderstädt U, Poppert S, Tannich E, Bull J, Kreikemeyer B, Barrantes I. Metagenomic Sequencing for the Diagnosis of Plasmodium spp. with Different Levels of Parasitemia in EDTA Blood of Malaria Patients—A Proof-of-Principle Assessment. International Journal of Molecular Sciences. 2022; 23(19):11150. https://doi.org/10.3390/ijms231911150
Chicago/Turabian StyleFrickmann, Hagen, Felix Weinreich, Ulrike Loderstädt, Sven Poppert, Egbert Tannich, Jana Bull, Bernd Kreikemeyer, and Israel Barrantes. 2022. "Metagenomic Sequencing for the Diagnosis of Plasmodium spp. with Different Levels of Parasitemia in EDTA Blood of Malaria Patients—A Proof-of-Principle Assessment" International Journal of Molecular Sciences 23, no. 19: 11150. https://doi.org/10.3390/ijms231911150
APA StyleFrickmann, H., Weinreich, F., Loderstädt, U., Poppert, S., Tannich, E., Bull, J., Kreikemeyer, B., & Barrantes, I. (2022). Metagenomic Sequencing for the Diagnosis of Plasmodium spp. with Different Levels of Parasitemia in EDTA Blood of Malaria Patients—A Proof-of-Principle Assessment. International Journal of Molecular Sciences, 23(19), 11150. https://doi.org/10.3390/ijms231911150