
Effect of Pt Decoration on the Optical Properties of Pristine and Defective MoS2: An Ab-Initio Study

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Pristine MoS2 Layer
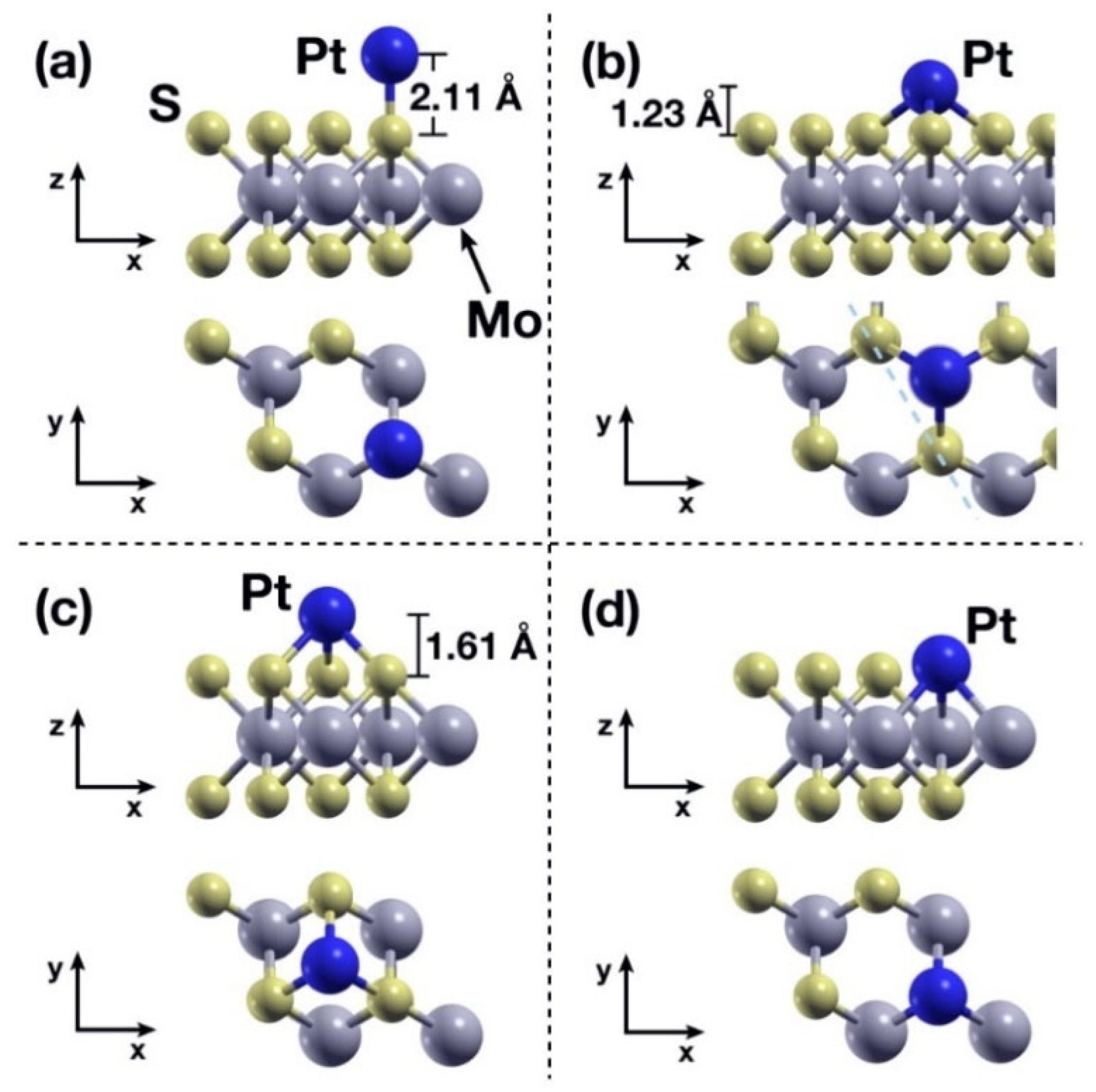
2.2. Static Calculations: Pt-Absorption on the MoS2 Layer
2.3. PDOS for the Pristine and Defective MoS2 Surfaces + Pt
2.4. Band Structures of the Pristine and Defective MoS2 Surfaces + Pt
2.5. Optical Properties
2.6. FPMD Calculation for the Weakest and Strongest Interactions
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Arellano, L.G.; De Santiago, F.; Miranda, Á.; Pérez, L.A.; Salazar, F.; Trejo, A.; Nakamura, J.; Cruz-Irisson, M. Ab Initio Study of Hydrogen Storage on Metal-Decorated GeC Monolayers. Int. J. Hydrogen Energy 2021, 46, 29261–29271. [Google Scholar] [CrossRef]
- Reddy, A.L.M.; Srivastava, A.; Gowda, S.R.; Gullapalli, H.; Dubey, M.; Ajayan, P.M. Synthesis Of Nitrogen-Doped Graphene Films For Lithium Battery Application. ACS Nano 2010, 4, 6337–6342. [Google Scholar] [CrossRef] [PubMed]
- Moschetto, S.; Bolognesi, M.; Prescimone, F.; Brucale, M.; Mezzi, A.; Ortolani, L.; Caporali, M.; Pingue, P.; Serrano-Ruiz, M.; Pisignano, D.; et al. Large-Area Oxidized Phosphorene Nanoflakes Obtained by Electrospray for Energy-Harvesting Applications. ACS Appl. Nano Mater. 2021, 4, 3476–3485. [Google Scholar] [CrossRef]
- Palmieri, V.; Perini, G.; De Spirito, M.; Papi, M. Graphene Oxide Touches Blood: In Vivo Interactions of Bio-Coronated 2D Materials. Nanoscale Horiz. 2019, 4, 273–290. [Google Scholar] [CrossRef]
- Donskyi, I.S.; Chen, Y.; Nickl, P.; Guday, G.; Qiao, H.; Achazi, K.; Lippitz, A.; Unger, W.E.S.; Böttcher, C.; Chen, W.; et al. Self-Degrading Graphene Sheets for Tumor Therapy. Nanoscale 2020, 12, 14222–14229. [Google Scholar] [CrossRef] [PubMed]
- Borandeh, S.; Alimardani, V.; Abolmaali, S.S.; Seppälä, J. Graphene Family Nanomaterials in Ocular Applications: Physicochemical Properties and Toxicity. Chem. Res. Toxicol. 2021, 34, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Liu, X.; Liu, F.; Du, G. First-Principles Based Ballistic Transport Simulation of Monolayer and Few-Layer InSe FETs. Jpn. J. Appl. Phys. 2019, 58, SBBA02. [Google Scholar] [CrossRef]
- Gonzalez-Medina, J.M.; Ruiz, F.G.; Marin, E.G.; Godoy, A.; Gámiz, F. Simulation Study of the Electron Mobility in Few-Layer MoS2 Metal–Insulator-Semiconductor Field-Effect Transistors. Solid-State Electron. 2015, 114, 30–34. [Google Scholar] [CrossRef]
- Wang, C.; Peng, L.; Wells, S.A.; Cain, J.D.; Huang, Y.-K.; Rhoads, L.A.; Dravid, V.P.; Hersam, M.C.; Grayson, M.A. Field-Effect Conductivity Scaling for Two-Dimensional Materials with Tunable Impurity Density. 2D Mater. 2022, 9, 031002. [Google Scholar] [CrossRef]
- Wu, H.; Cui, Y.; Xu, J.; Yan, Z.; Xie, Z.; Hu, Y.; Zhu, S. Multifunctional Half-Floating-Gate Field-Effect Transistor Based on MoS 2 –BN–Graphene van Der Waals Heterostructures. Nano Lett. 2022, 22, 2328–2333. [Google Scholar] [CrossRef]
- Sosa, A.N.; Cid, B.J.; Hernández-Hernández, I.J.; Miranda, Á. Effects of Substitutional Doping and Vacancy Formation on the Structural and Electronic Properties of Siligene: A DFT Study. Mater. Lett. 2022, 307, 130993. [Google Scholar] [CrossRef]
- Pessanha, T.M.; Paschoalino, W.J.; Deroco, P.B.; Kogikoski, S.; De Moraes, A.C.M.; Carvalho Castro Silva, C.; Kubota, L.T. Interfacial Capacitance of Graphene Oxide Films Electrodes: Fundamental Studies on Electrolytes Interface Aiming (Bio)Sensing Applications. Electroanalysis 2022, 34, 692–700. [Google Scholar] [CrossRef]
- Dickinson, R.G.; Pauling, L. The Crystal Structure of Molybdenite. J. Am. Chem. Soc. 1923, 45, 1466–1471. [Google Scholar] [CrossRef]
- Schönfeld, B.; Huang, J.J.; Moss, S.C. Anisotropic mean-square displacements (MSD) in single-crystals of 2H-and 3R-MoS2. Acta Crystallogr. Sect. B Struct. Sci. 1983, 39, 404–407. [Google Scholar] [CrossRef]
- Gupta, A.; Sakthivel, T.; Seal, S. Recent Development in 2D Materials beyond Graphene. Prog. Mater. Sci. 2015, 73, 44–126. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhong, L.; Wang, H.; Li, X. Structural Defects, Mechanical Behaviors, and Properties of Two-Dimensional Materials. Materials 2021, 14, 1192. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, S.B.; Zheng, J.; Du, J.; Wen, S.C.; Tang, D.Y.; Loh, K.P. Molybdenum Disulfide (MoS_2) as a Broadband Saturable Absorber for Ultra-Fast Photonics. Opt. Express 2014, 22, 7249. [Google Scholar] [CrossRef]
- Yin, W.; Yan, L.; Yu, J.; Tian, G.; Zhou, L.; Zheng, X.; Zhang, X.; Yong, Y.; Li, J.; Gu, Z.; et al. High-Throughput Synthesis of Single-Layer MoS2 Nanosheets as a Near-Infrared Photothermal-Triggered Drug Delivery for Effective Cancer Therapy. ACS Nano 2014, 8, 6922–6933. [Google Scholar] [CrossRef]
- Liang, W.; Luo, X. Theoretical Studies of MoS 2 and Phosphorene Drug Delivery for Antituberculosis Drugs. J. Phys. Chem. C 2020, 124, 8279–8287. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Cheng, Y.; Cao, L.; Bai, F.; Yue, S.; Xie, P.; Ma, J. Molybdenum Disulfide (MoS2): A Novel Activator of Peracetic Acid for the Degradation of Sulfonamide Antibiotics. Water Res. 2021, 201, 117291. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Yang, L.; Zhao, G.; Zhou, H.; Cai, F.; Li, Y.; Wang, X.; Yu, F. Boosting pH-Universal Hydrogen Evolution of Molybdenum Disulfide Particles by Interfacial Engineering †. Chin. J. Chem. 2021, 39, 288–294. [Google Scholar] [CrossRef]
- Liaqat, A.; Yin, Y.; Hussain, S.; Wen, W.; Wu, J.; Guo, Y.; Dang, C.; Ho, C.-H.; Liu, Z.; Yu, P.; et al. An All Two-Dimensional Vertical Heterostructure Graphene/CuInP2S6/MoS2 for Negative Capacitance Field Effect Transistor. Nanotechnology 2022, 33, 125703. [Google Scholar] [CrossRef] [PubMed]
- Vatalaro, M.; De Rose, R.; Lanuzza, M.; Magnone, P.; Conti, S.; Iannaccone, G.; Crupi, F. Assessment of Paper-Based MoS2 FET for Physically Unclonable Functions. Solid-State Electron. 2022, 194, 108391. [Google Scholar] [CrossRef]
- Espejo, C.; Rangel, T.; Romero, A.H.; Gonze, X.; Rignanese, G.-M. Band Structure Tunability in MoS 2 under Interlayer Compression: A DFT and GW Study. Phys. Rev. B 2013, 87, 245114. [Google Scholar] [CrossRef]
- Zhou, W.; Zou, X.; Najmaei, S.; Liu, Z.; Shi, Y.; Kong, J.; Lou, J.; Ajayan, P.M.; Yakobson, B.I.; Idrobo, J.-C. Intrinsic Structural Defects in Monolayer Molybdenum Disulfide. Nano Lett. 2013, 13, 2615–2622. [Google Scholar] [CrossRef]
- Lee, J.G. Computational Materials Science: An Introduction, 2nd ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2017; ISBN 978-1-4987-4973-2. [Google Scholar]
- Oura, K. (Ed.) Surface Science: An Introduction; Advanced Texts in Physics; Springer: Berlin, Germany; New York, NY, USA, 2003; ISBN 978-3-540-00545-2. [Google Scholar]
- Patel, M.; Chavda, A.; Mukhopadhyay, I.; Kim, J.; Ray, A. Nanostructured SnS with Inherent Anisotropic Optical Properties for High Photoactivity. Nanoscale 2016, 8, 2293–2303. [Google Scholar] [CrossRef]
- Viezbicke, B.D.; Patel, S.; Davis, B.E.; Birnie, D.P. Evaluation of the Tauc Method for Optical Absorption Edge Determination: ZnO Thin Films as a Model System: Tauc Method for Optical Absorption Edge Determination. Phys. Status Solidi 2015, 252, 1700–1710. [Google Scholar] [CrossRef]
- Jiao, S.; Kong, M.; Hu, Z.; Zhou, S.; Xu, X.; Liu, L. Pt Atom on the Wall of Atomic Layer Deposition (ALD)-Made MoS 2 Nanotubes for Efficient Hydrogen Evolution. Small 2022, 18, 2105129. [Google Scholar] [CrossRef]
- Park, J.; Mun, J.; Shin, J.-S.; Kang, S.-W. Highly Sensitive Two-Dimensional MoS 2 Gas Sensor Decorated with Pt Nanoparticles. R. Soc. Open Sci. 2018, 5, 181462. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Zhang, C.; Ji, J.; Jin, P.; Zhou, X. Facile, Rapid, and Well-Controlled Preparation of Pt Nanoparticles Decorated on Single Surface of Mos 2 Nanosheets and Application in HER. ChemNanoMat 2020, 6, 435–441. [Google Scholar] [CrossRef]
- Ren, W.; Zhang, H.; Cheng, C. Ultrafine Pt Nanoparticles Decorated MoS 2 Nanosheets with Significantly Improved Hydrogen Evolution Activity. Electrochim. Acta 2017, 241, 316–322. [Google Scholar] [CrossRef]
- Burman, D.; Santra, S.; Pramanik, P.; Guha, P.K. Pt Decorated MoS 2 Nanoflakes for Ultrasensitive Resistive Humidity Sensor. Nanotechnology 2018, 29, 115504. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Chapter 3: Crystal Binding and Elastic Constants. In Introduction to Solid State Physics; Wiley: Hoboken, NJ, USA, 2005; p. 50. ISBN 978-0-471-41526-8. [Google Scholar]
- Yang, Y.; Zhang, Y.; Ye, H.; Yu, Z.; Liu, Y.; Su, B.; Xu, W. Structural and Electronic Properties of 2H Phase Janus Transition Metal Dichalcogenide Bilayers. Superlattices Microstruct. 2019, 131, 8–14. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Espresso: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced Capabilities for Materials Modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in Phys. Rev. Lett. 1997, 78, 1396–1396. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Computational science series; Academic Press: San Diego, CA, USA, 2002; ISBN 978-0-12-267351-1. [Google Scholar]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Theory and Implementation. In Modern Methods and Algorithms of Quantum Chemistry, Proceedings; Grotendorst, J., Ed.; NIC Series; John von Neumann Institute for Computing: Jülich, Germany, 2000; Volume 3, pp. 329–477. ISBN 3-00-005834-6. [Google Scholar]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, 1st ed.; Cambridge University Press: Cambridge, UK, 2012; ISBN 978-1-107-66353-4. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Kokalj, A. XCrySDen—A New Program for Displaying Crystalline Structures and Electron Densities. J. Mol. Graph. Model. 1999, 17, 176–179. [Google Scholar] [CrossRef]
- Lucarini, V. (Ed.) Kramers-Kronig Relations in Optical Materials Research; Springer Series in Optical Sciences; Springer: Berlin, Germany; New York, NY, USA, 2005; ISBN 978-3-540-23673-3. [Google Scholar]
- Jiménez-González, A.F.; Ramírez-de-Arellano, J.M.; Magaña, L.F. Substantial Variations in the Optical Absorption and Reflectivity of Graphene When the Concentrations of Vacancies and Doping with Fluorine, Nitrogen, and Oxygen Change. Int. J. Mol. Sci. 2021, 22, 6832. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case 1 | Description | Eads |
|---|---|---|
| a | Pt over S, pristine MoS2 | −2.77 |
| b | Pt over S-S bond, pristine MoS2 | −3.45 |
| c | Pt over S-S-S triangle, pristine MoS2 | −2.94 |
| d | Pt over VS vacancy | −5.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-de-Arellano, J.M.; Jiménez-González, A.F.; Canales, M.; Magaña, L.F. Effect of Pt Decoration on the Optical Properties of Pristine and Defective MoS2: An Ab-Initio Study. Int. J. Mol. Sci. 2022, 23, 11199. https://doi.org/10.3390/ijms231911199
Ramírez-de-Arellano JM, Jiménez-González AF, Canales M, Magaña LF. Effect of Pt Decoration on the Optical Properties of Pristine and Defective MoS2: An Ab-Initio Study. International Journal of Molecular Sciences. 2022; 23(19):11199. https://doi.org/10.3390/ijms231911199
Chicago/Turabian StyleRamírez-de-Arellano, Juan Manuel, Ali Fransuani Jiménez-González, Mónica Canales, and Luis Fernando Magaña. 2022. "Effect of Pt Decoration on the Optical Properties of Pristine and Defective MoS2: An Ab-Initio Study" International Journal of Molecular Sciences 23, no. 19: 11199. https://doi.org/10.3390/ijms231911199
APA StyleRamírez-de-Arellano, J. M., Jiménez-González, A. F., Canales, M., & Magaña, L. F. (2022). Effect of Pt Decoration on the Optical Properties of Pristine and Defective MoS2: An Ab-Initio Study. International Journal of Molecular Sciences, 23(19), 11199. https://doi.org/10.3390/ijms231911199