
Polo-Like Kinase 2 Plays an Essential Role in Cytoprotection against MG132-Induced Proteasome Inhibition via Phosphorylation of Serine 19 in HSPB5

 ,
,
Abstract
:1. Introduction
2. Results
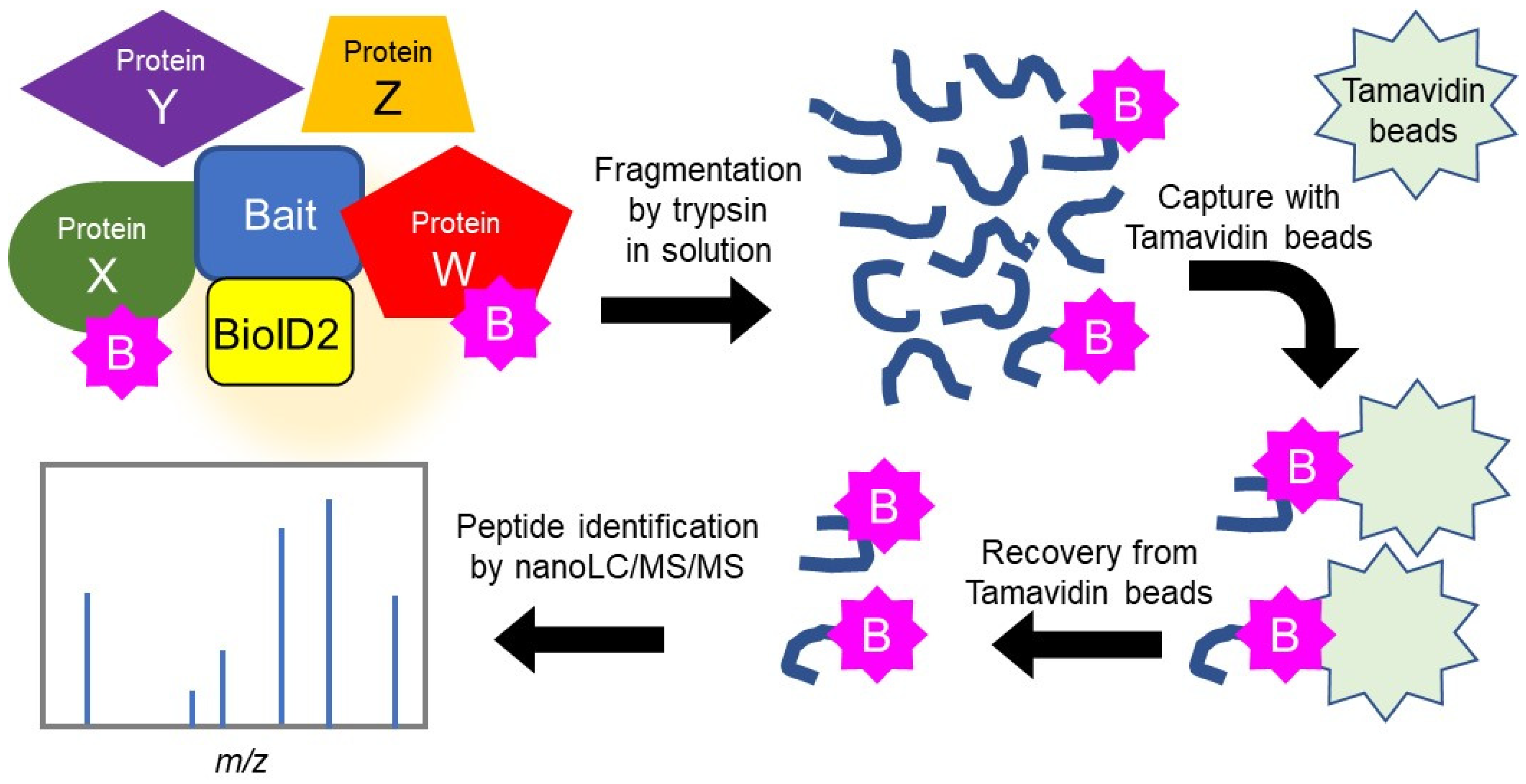
2.1. Search for HSPB5 Binding Proteins by Proximity-Dependent Biotin Labeling
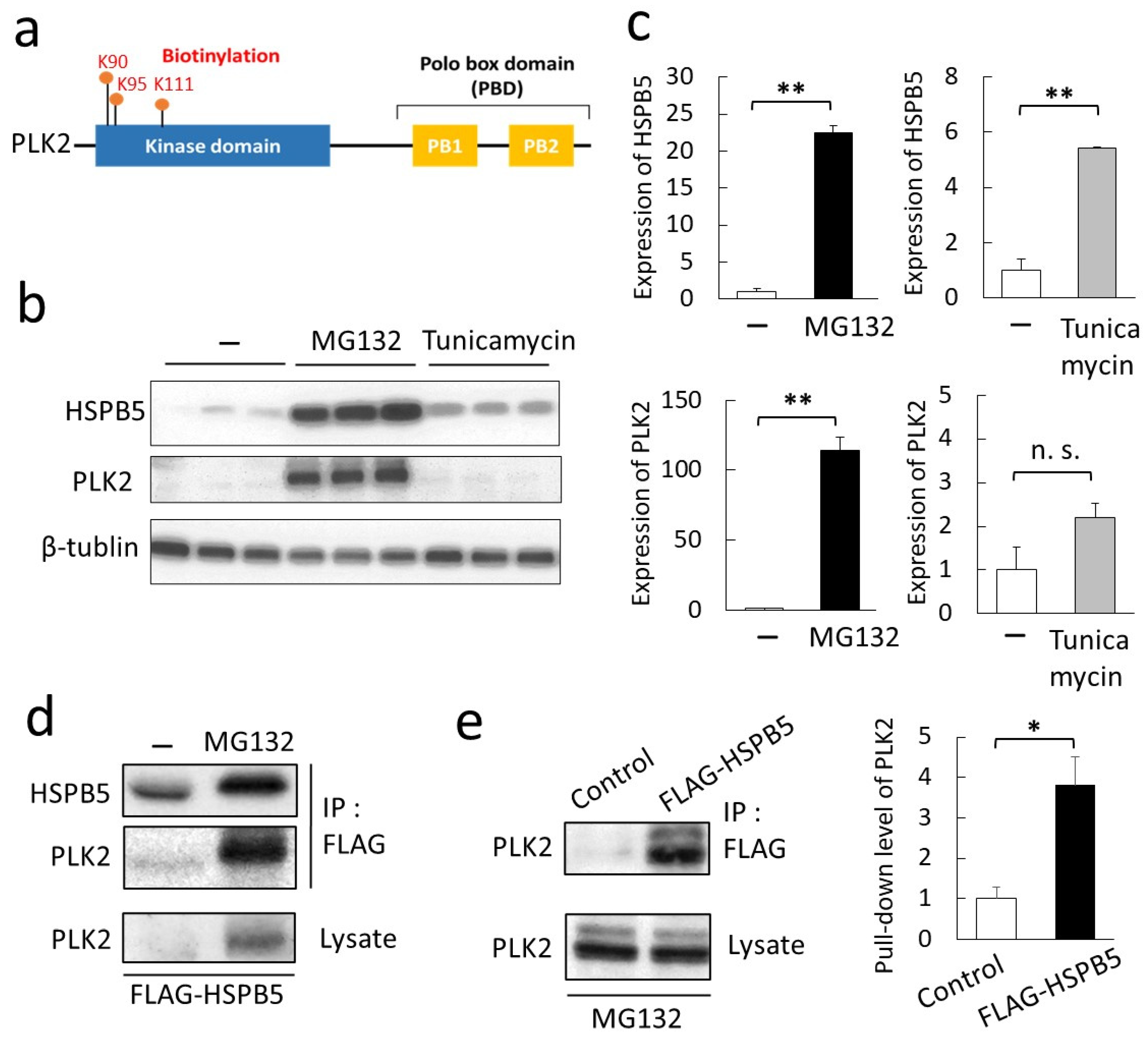
2.2. Validation of a New Binding Protein, PLK2
2.3. Phosphorylation of HSPB5 by PLK2 Triggered by ER Stress via Proteasome Inhibition
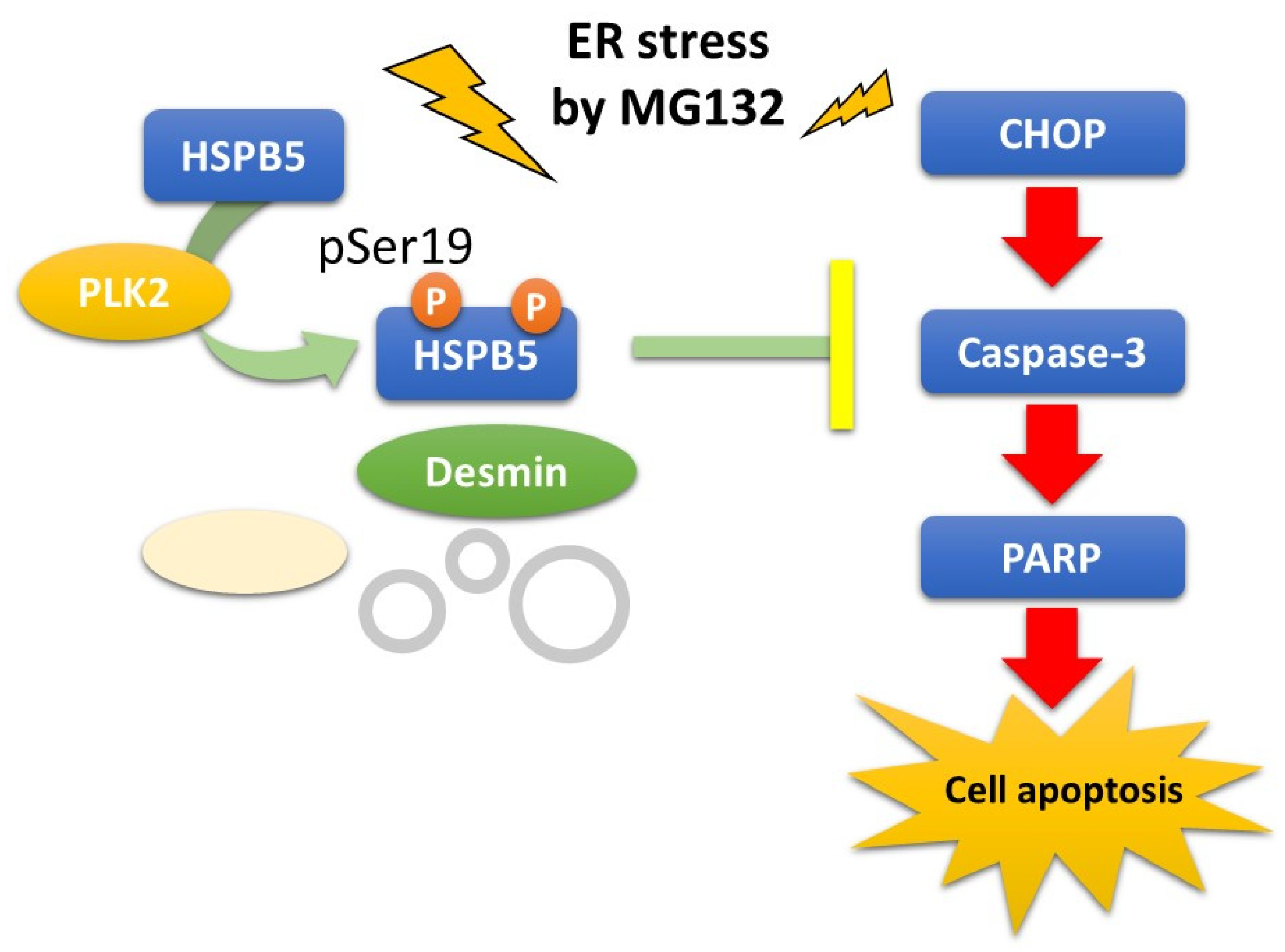
2.4. Critical Role of HSPB5 and PLK2 in ER Stress Response via Proteasome Inhibition
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Plasmid Construction
4.3. Cell Culture
4.4. Identification of Binding Proteins Using the BioID Method
4.5. Western Blot Analysis
4.6. HSPB5 Binding Assay
4.7. Immunofluorescence Microscopy
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| emPAI | exponentially modified protein abundance index |
| PLK2 | polo-like kinase 2 |
| CHOP | C/EBP homologous protein |
| siRNA | small interference RNA |
| BioID | proximity-dependent biotin identification |
| nanoLC/MS/MS | nanoscale liquid chromatography coupled to tandem mass spectrometry |
| MK2 | MAP kinase-activated protein kinase 2 |
| p38 | p38 MAP kinases |
| siRNA | small interference RNA |
| TFA | trifluoroacetic acid |
| DAPI | 6-diamidino-2-phenylindole |
| PARP | poly ADP-ribose polymerase |
References
- Frankowska, N.; Lisowska, K.; Witkowski, J.M. Proteolysis dysfunction in the process of aging and age-related diseases. Front. Aging 2022, 3, 927630. [Google Scholar] [CrossRef] [PubMed]
- Afroze, D.; Kumar, A. ER stress in skeletal muscle remodeling and myopathies. FEBS J. 2019, 286, 379–398. [Google Scholar] [CrossRef] [PubMed]
- Gallot, Y.S.; Bohnert, K.R. Confounding Roles of ER Stress and the Unfolded Protein Response in Skeletal Muscle Atrophy. Int. J. Mol. Sci. 2021, 22, 2567. [Google Scholar] [CrossRef] [PubMed]
- Zito, E. Targeting ER stress/ER stress response in myopathies. Redox Biol. 2019, 26, 101232. [Google Scholar] [CrossRef]
- Sun-Wang, J.L.; Ivanova, S.; Zorzano, A. The dialogue between the ubiquitin-proteasome system and autophagy: Implications in ageing. Ageing Res. Rev. 2020, 64, 101203. [Google Scholar] [CrossRef]
- Hayashi, J.; Carver, J.A. The multifaceted nature of αB-crystallin. Cell Stress Chaperon 2020, 25, 639–654. [Google Scholar] [CrossRef]
- Brady, J.P.; Garland, D.L.; Green, D.E.; Tamm, E.R.; Giblin, F.J.; Wawrousek, E.F. αB-Crystallin in Lens Development and Muscle Integrity: A Gene Knockout Approach. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2924–2934. [Google Scholar]
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H.; Teraoka, K.; Chikamori, T.; Yamashina, A.; Kimura, A. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386. [Google Scholar] [CrossRef]
- Ueda, S.; Kokaji, Y.; Simizu, S.; Honda, K.; Yoshino, K.; Kamisoyama, H.; Shirai, Y.; Yamanoue, M. Chicken heat shock protein HSPB1 increases and interacts with αB-crystallin in aged skeletal muscle. Biosci. Biotechnol. Biochem. 2015, 79, 1867–1875. [Google Scholar] [CrossRef]
- Bakthisaran, R.; Akula, K.K.; Tangirala, R.; Rao, C.M. Phosphorylation of αB-crystallin: Role in stress, aging and patho-physiological conditions. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Muranova, L.K.; Sudnitsyna, M.V.; Gusev, N.B. αB-Crystallin Phosphorylation: Advances and Problems. Biochem. (Mosc.) 2018, 83, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Dimitrova, V.; Gibert, B.; Virot, S.; Mounier, N.; Nivon, M.; Kretz-Remy, C.; Corset, V.; Mehlen, P.; Arrigo, A.-P. Analysis of the dominant effects mediated by wild type or R120G mutant of αB-crystallin (HspB5) towards Hsp27 (HspB1). PLoS ONE 2013, 8, e70545. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.M.; May, D.G.; Roux, K.J. BioID as a Tool for Protein-Proximity Labeling in Living Cells. Methods Mol. Biol. 2019, 2012, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Kido, K.; Yamanaka, S.; Nakano, S.; Motani, K.; Shinohara, S.; Nozawa, A.; Kosako, H.; Ito, S.; Sawasaki, T. AirID, a novel proximity biotinylation enzyme, for analysis of protein-protein interactions. eLife 2020, 9, e54983. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Blee, A.M.; Macway, K.G.; Renner, D.J.; Yamada, S. Force dependent biotinylation of myosin IIA by α-catenin tagged with a promiscuous biotin ligase. PLoS ONE 2015, 10, e0122886. [Google Scholar] [CrossRef]
- Cheah, J.S.; Jacobs, K.A.; Lai, T.W.; Caballelo, R.; Yee, J.L.; Ueda, S.; Heinrich, V.; Yamada, S. Spatial proximity of proteins surrounding zyxin under force-bearing conditions. Mol. Biol. Cell 2021, 32, 1221–1228. [Google Scholar] [CrossRef]
- Fribley, A.; Wang, C.Y. Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol. Ther. 2006, 5, 745–748. [Google Scholar] [CrossRef]
- Doran, P.; Donoghue, P.; O’Connell, K.; Gannon, J.; Ohlendieck, K. Proteomics of skeletal muscle aging. Proteomics 2009, 9, 989–1003. [Google Scholar] [CrossRef]
- Lin, I.H.; Chang, J.-L.; Hua, K.; Huang, W.-C.; Hsu, M.-T.; Chen, Y.-F. Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet. 2018, 19, 55. [Google Scholar] [CrossRef]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiapparelli, L.M.; McClatchy, D.B.; Liu, H.H.; Sharma, P.; Yates, J.R., 3rd; Cline, H.T. Direct detection of biotinylated proteins by mass spectrometry. J. Proteome Res. 2014, 13, 3966–3978. [Google Scholar] [CrossRef] [PubMed]
- Motani, K.; Kosako, H. BioID screening of biotinylation sites using the avidin-like protein Tamavidin 2-REV identifies global interactors of stimulator of interferon genes (STING). J. Biol. Chem. 2020, 295, 11174–11183. [Google Scholar] [CrossRef] [PubMed]
- Kressin, M.; Fietz, D.; Becker, S.; Strebhardt, K. Modelling the Functions of Polo-Like Kinases in Mice and Their Applications as Cancer Targets with a Special Focus on Ovarian Cancer. Cells 2021, 10, 1176. [Google Scholar] [CrossRef]
- Kisselev, A.F. Site-Specific Proteasome Inhibitors. Biomolecules 2021, 12, 54. [Google Scholar] [CrossRef]
- Golenhofen, N.; Perng, M.D.; Quinlan, R.A.; Drenckhahn, D. Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem. Cell Biol. 2004, 122, 415–425. [Google Scholar] [CrossRef]
- Johnson, E.F.; Stewart, K.D.; Woods, K.W.; Giranda, V.L.; Luo, Y. Pharmacological and Functional Comparison of the Polo-like Kinase Family: Insight into Inhibitor and Substrate Specificity. Biochemistry 2007, 46, 9551–9563. [Google Scholar] [CrossRef]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prévost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tomé, F.; Dupret, J.M.; et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Agnetti, G.; Herrmann, H.; Cohen, S. New roles for desmin in the maintenance of muscle homeostasis. FEBS J. 2021, 289, 2755–2770. [Google Scholar] [CrossRef]
- Tidball, J.G.; Spencer, M.J. PDGF stimulation induces phosphorylation of talin and cytoskeletal reorganization in skeletal muscle. J. Cell Biol. 1993, 123, 627–635. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Jubin, T.; Kadam, A.; Jariwala, M.; Bhatt, S.; Sutariya, S.; Gani, A.R.; Gautam, S.; Begum, R. The PARP family: Insights into functional aspects of poly (ADP-ribose) polymerase-1 in cell growth and survival. Cell Prolif. 2016, 49, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Dimauro, I.; Caporossi, D. Alpha B-Crystallin in Muscle Disease Prevention: The Role of Physical Activity. Molecules 2022, 27, 1147. [Google Scholar] [CrossRef]
- Cox, D.; Ecroyd, H. The small heat shock proteins αB-crystallin (HSPB5) and Hsp27 (HSPB1) inhibit the intracellular aggregation of α-synuclein. Cell Stress Chaperon. 2017, 22, 589–600. [Google Scholar] [CrossRef]
- Sanbe, A.; Daicho, T.; Mizutani, R.; Endo, T.; Miyauchi, N.; Yamauchi, J.; Tanonaka, K.; Glabe, C.; Tanoue, A. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS ONE 2009, 4, e5351. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Nishimura, R.N.; Sharp, F.R. Heat shock proteins and neuromuscular disease. Muscle Nerve 2005, 32, 693–709. [Google Scholar] [CrossRef]
- Muraleva, N.A.; Kolosova, N.G.; Stefanova, N.A. p38 MAPK–dependent alphaB-crystallin phosphorylation in Alzheimer’s disease–like pathology in OXYS rats. Experimental Gerontology 2019, 119, 45–52. [Google Scholar] [CrossRef]
- Shen, T.; Li, Y.; Chen, Z.; Liang, S.; Guo, Z.; Wang, P.; Wu, Q.; Ba, G.; Fu, Q. CHOP negatively regulates Polo-like kinase 2 expression via recruiting C/EBPα to the upstream-promoter in human osteosarcoma cell line during ER stress. Int. J. Biochem. Cell Biol. 2017, 89, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, K.; Van Itallie, C.M.; Aponte, A.; Gucek, M.; Tietgens, A.J.; Anderson, J.M. Proteomic analysis of proteins surrounding occludin and claudin-4 reveals their proximity to signaling and trafficking networks. PLoS ONE 2015, 10, e0117074. [Google Scholar] [CrossRef] [PubMed]
- Franchin, C.; Cesaro, L.; Pinna, L.A.; Arrigoni, G.; Salvi, M. Identification of the PLK2-dependent phosphopeptidome by quantitative proteomics [corrected]. PLoS ONE 2014, 9, e111018. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Staszewska, I.; Mihailovska, E.; Fischer, I.; Goldmann, W.; Schröder, R.; Wiche, G. Chemical chaperone ameliorates pathological protein aggregation in plectin-deficient muscle. J. Clin. Investig. 2014, 124, 1144–1157. [Google Scholar] [CrossRef]
- Bartelt-Kirbach, B.; Wiegreffe, C.; Birk, S.; Baur, T.; Moron, M.; Britsch, S.; Golenhofen, N. HspB5/αB-crystallin phosphorylation at S45 and S59 is essential for protection of the dendritic tree of rat hippocampal neurons. J. Neurochem. 2021, 157, 2055–2069. [Google Scholar] [CrossRef]
- Ecroyd, H.; Meehan, S.; Horwitz, J.; Aquilina, J.A.; Benesch, J.L.P.; Robinson, C.V.; Macphee, C.E.; Carver, J.A. Mimicking phosphorylation of alphaB-crystallin affects its chaperone activity. Biochem. J. 2007, 401, 129–141. [Google Scholar] [CrossRef]
- Morrison, L.E.; Hoover, H.E.; Thuerauf, D.J.; Glembotski, C.C. Mimicking Phosphorylation of αB-Crystallin on Serine-59 Is Necessary and Sufficient to Provide Maximal Protection of Cardiac Myocytes From Apoptosis. Circ. Res. 2003, 92, 203–211. [Google Scholar] [CrossRef]
- Ueda, S.; Kataoka, T.; Satoh, T. Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol. Cell 2008, 100, 645–657. [Google Scholar] [CrossRef]
- Hidese, R.; Toyoda, M.; Yoshino, K.I.; Fukuda, W.; Wihardja, G.A.; Kimura, S.; Fujita, J.; Niitsu, M.; Oshima, T.; Imanaka, T.; et al. The C-terminal flexible region of branched-chain polyamine synthase facilitates substrate specificity and catalysis. FEBS J. 2019, 286, 3926–3940. [Google Scholar] [CrossRef]
- Ueda, S.; Hosoda, M.; Kasamatsu, K.; Horiuchi, M.; Nakabayashi, R.; Kang, B.; Shinohara, M.; Nakanishi, H.; Ohto-Nakanishi, T.; Yamanoue, M.; et al. Production of Hydroxy Fatty Acids, Precursors of γ-Hexalactone, Contributes to the Characteristic Sweet Aroma of Beef. Metabolites 2022, 12, 332. [Google Scholar] [CrossRef]
- Ueda, S.; Hosoda, M.; Yoshino, K.-i.; Yamanoue, M.; Shirai, Y. Gene Expression Analysis Provides New Insights into the Mechanism of Intramuscular Fat Formation in Japanese Black Cattle. Genes 2021, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Takashima, Y.; Gotou, Y.; Sasaki, R.; Nakabayashi, R.; Suzuki, T.; Sasazaki, S.; Fukuda, I.; Kebede, B.; Kadowaki, Y.; et al. Application of Mass Spectrometry for Determining the Geographic Production Area of Wagyu Beef. Metabolites 2022, 12, 777. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| emPAI a | ||
|---|---|---|
| Protein Name | MG132 (−) | MG132 (+) |
| PHD finger-like domain-containing protein 5A | 0 | 0.26 |
| Poly(rC)-binding protein 2 | 0 | 0.18 |
| Translocon-associated protein subunit beta | 0 | 0.17 |
| Dynactin subunit 2 | 0 | 0.15 |
| Serine/threonine-protein kinase PLK2 | 0 | 0.13 |
| 6-phosphogluconate dehydrogenase, | 0 | 0.13 |
| ADP-ribosylation factor GTPase-activating protein 2 | 0 | 0.12 |
| Pyruvate kinase PKM | 0.06 | 0.18 |
| Paired mesoderm homeobox protein 1 | 0 | 0.12 |
| Eukaryotic initiation factor 4A-I | 0.07 | 0.15 |
| Phosphoglycerate kinase 1 | 0 | 0.07 |
| 26S proteasome regulatory subunit 6A | 0 | 0.07 |
| Sequestosome-1 | 0.07 | 0.14 |
| Abl interactor 1 | 0 | 0.06 |
| PDZ and LIM domain protein 7 | 0 | 0.06 |
| Heat shock cognate 71 kDa protein | 0 | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, S.; Nishihara, M.; Hioka, Y.; Yoshino, K.-i.; Yamada, S.; Yamanoue, M.; Shirai, Y. Polo-Like Kinase 2 Plays an Essential Role in Cytoprotection against MG132-Induced Proteasome Inhibition via Phosphorylation of Serine 19 in HSPB5. Int. J. Mol. Sci. 2022, 23, 11257. https://doi.org/10.3390/ijms231911257
Ueda S, Nishihara M, Hioka Y, Yoshino K-i, Yamada S, Yamanoue M, Shirai Y. Polo-Like Kinase 2 Plays an Essential Role in Cytoprotection against MG132-Induced Proteasome Inhibition via Phosphorylation of Serine 19 in HSPB5. International Journal of Molecular Sciences. 2022; 23(19):11257. https://doi.org/10.3390/ijms231911257
Chicago/Turabian StyleUeda, Shuji, Moeka Nishihara, Yuuki Hioka, Ken-ichi Yoshino, Soichiro Yamada, Minoru Yamanoue, and Yasuhito Shirai. 2022. "Polo-Like Kinase 2 Plays an Essential Role in Cytoprotection against MG132-Induced Proteasome Inhibition via Phosphorylation of Serine 19 in HSPB5" International Journal of Molecular Sciences 23, no. 19: 11257. https://doi.org/10.3390/ijms231911257
APA StyleUeda, S., Nishihara, M., Hioka, Y., Yoshino, K. -i., Yamada, S., Yamanoue, M., & Shirai, Y. (2022). Polo-Like Kinase 2 Plays an Essential Role in Cytoprotection against MG132-Induced Proteasome Inhibition via Phosphorylation of Serine 19 in HSPB5. International Journal of Molecular Sciences, 23(19), 11257. https://doi.org/10.3390/ijms231911257