
RUNX1/CEBPA Mutation in Acute Myeloid Leukemia Promotes Hypermethylation and Indicates for Demethylation Therapy
 ,
,  and
and
Abstract
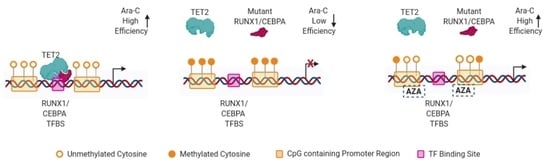
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
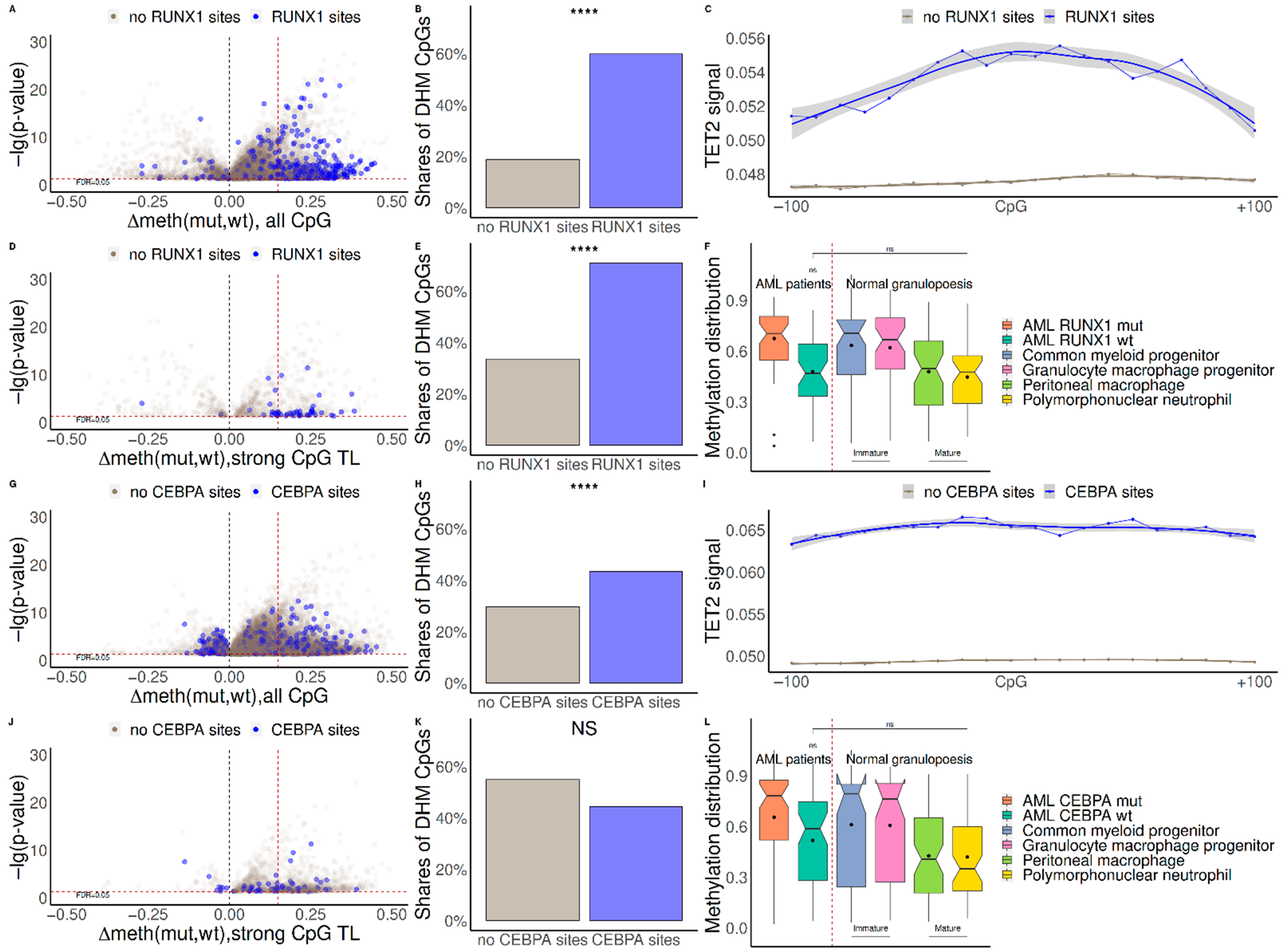
2.1. Mutations in RUNX1 and CEBPA Lead to Hypermethylation near Their Binding Sites and Change of Expression of Regulated Genes
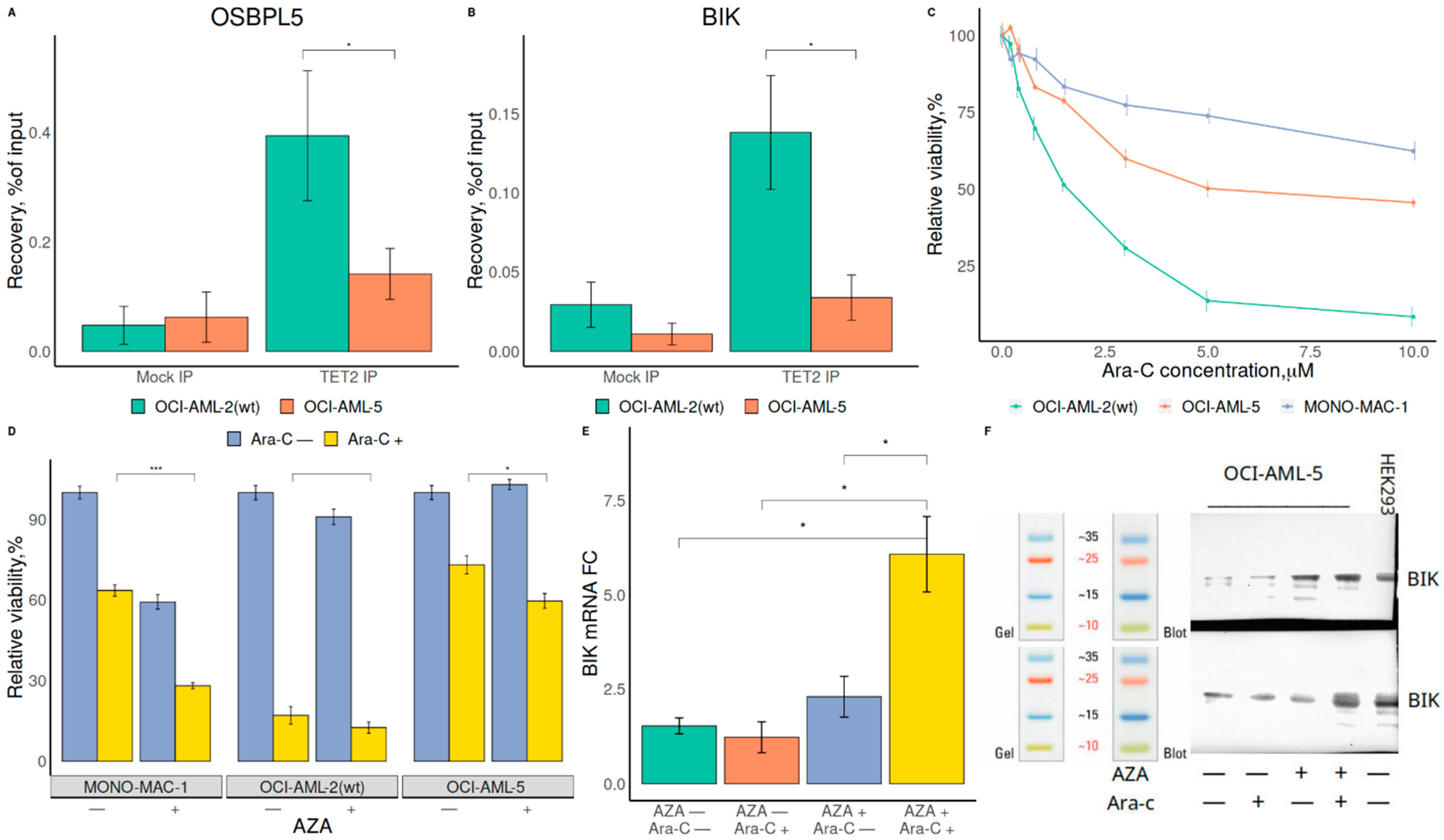
2.2. TET2 Is Likely Involved in RUNX1/CEBPA TFBS Demethylation in AML
2.3. Demethylation Treatment Restores the Sensitivity of RUNX1-Mutated AML Cells to Ara-C
3. Discussion
4. Materials and Methods
4.1. Patients Data
4.2. Additional Data
4.3. TFBS Prediction
4.4. Statistical Analysis
4.5. ChIP-qPCR
4.6. Cell Treatment
4.7. Cell Viability Testing
4.8. Western Blots
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute Myeloid Leukemia. |
| CpG TL | CpG Traffic Lights. |
| Strong CpG TL | CpG traffic lights which are significantly differentially expressed and methylated. |
| DEG | Differentially expressed genes. |
| DM CpG | Differentially methylated CpG. |
| DHM CpG | Differently hypermethylated CpG. |
| SCC | Spearman Correlation Coefficient. |
| SDS | Sodium Dodecyl Sulfate. |
| TCGA | The Cancer Genome Atlas. |
| TF | Transcription Factors. |
| TFBS | Transcription Factor Binding Sites. |
References
- Nimer, S.D. Is it important to decipher the heterogeneity of “normal karyotype AML”? Best Pract. Res. Clin. Haematol. 2008, 21, 43–52. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Conway O’Brien, E.; Prideaux, S.; Chevassut, T. The epigenetic landscape of acute myeloid leukemia. Adv. Hematol. 2014, 2014, 103175. [Google Scholar] [CrossRef]
- Gardin, C.; Dombret, H. Hypomethylating Agents as a Therapy for AML. Curr. Hematol. Malig. Rep. 2017, 12, 1–10. [Google Scholar] [CrossRef]
- Rowe, J.M. Progress and predictions: AML in 2018. Best Pract. Res. Clin. Haematol. 2018, 31, 337–340. [Google Scholar] [CrossRef]
- Martelli, M.P.; Martino, G.; Cardinali, V.; Falini, B.; Martinelli, G.; Cerchione, C. Enasidenib and ivosidenib in AML. Minerva Med. 2020, 111, 411–426. [Google Scholar] [CrossRef]
- Ichikawa, M.; Yoshimi, A.; Nakagawa, M.; Nishimoto, N.; Watanabe-Okochi, N.; Kurokawa, M. A role for RUNX1 in hematopoiesis and myeloid leukemia. Int. J. Hematol. 2013, 97, 726–734. [Google Scholar] [CrossRef]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J. Clin. Oncol. 2012, 30, 3109–3118. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Röck, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. 2011, 29, 1364–1372. [Google Scholar] [CrossRef]
- Tang, J.-L.; Hou, H.-A.; Chen, C.-Y.; Liu, C.-Y.; Chou, W.-C.; Tseng, M.-H.; Huang, C.-F.; Lee, F.-Y.; Liu, M.-C.; Yao, M.; et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: Prognostic implication and interaction with other gene alterations. Blood 2009, 114, 5352–5361. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood 2011, 117, 2348–2357. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Köhne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168. [Google Scholar] [CrossRef]
- Hasemann, M.S.; Lauridsen, F.K.B.; Waage, J.; Jakobsen, J.S.; Frank, A.-K.; Schuster, M.B.; Rapin, N.; Bagger, F.O.; Hoppe, P.S.; Schroeder, T.; et al. C/EBPα is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet. 2014, 10, e1004079. [Google Scholar] [CrossRef]
- Fröhling, S.; Schlenk, R.F.; Stolze, I.; Bihlmayr, J.; Benner, A.; Kreitmeier, S.; Tobis, K.; Döhner, H.; Döhner, K. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: Prognostic relevance and analysis of cooperating mutations. J. Clin. Oncol. 2004, 22, 624–633. [Google Scholar] [CrossRef]
- Dufour, A.; Schneider, F.; Metzeler, K.H.; Hoster, E.; Schneider, S.; Zellmeier, E.; Benthaus, T.; Sauerland, M.-C.; Berdel, W.E.; Büchner, T.; et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J. Clin. Oncol. 2010, 28, 570–577. [Google Scholar] [CrossRef]
- Pastore, F.; Kling, D.; Hoster, E.; Dufour, A.; Konstandin, N.P.; Schneider, S.; Sauerland, M.C.; Berdel, W.E.; Buechner, T.; Woermann, B.; et al. Long-term follow-up of cytogenetically normal CEBPA-mutated AML. J. Hematol. Oncol. 2014, 7, 55. [Google Scholar] [CrossRef]
- Wouters, B.J.; Löwenberg, B.; Erpelinck-Verschueren, C.A.J.; van Putten, W.L.J.; Valk, P.J.M.; Delwel, R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009, 113, 3088–3091. [Google Scholar] [CrossRef]
- Tarlock, K.; Lamble, A.J.; Wang, Y.-C.; Gerbing, R.B.; Ries, R.E.; Loken, M.R.; Brodersen, L.E.; Pardo, L.; Leonti, A.; Smith, J.L.; et al. CEBPA-bZip mutations are associated with favorable prognosis in de novo AML: A report from the Children’s Oncology Group. Blood 2021, 138, 1137–1147. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, M.; Chen, X.; Chen, L.; Xu, Y.; Lv, L.; Wang, P.; Yang, H.; Ma, S.; Lin, H.; et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 2015, 57, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Maeda, S.; Furuhata, E.; Shimizu, Y.; Nishimura, H.; Kishima, M.; Suzuki, H. A screening system to identify transcription factors that induce binding site-directed DNA demethylation. Epigenetics Chromatin 2017, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shimizu, Y.; Furuhata, E.; Maeda, S.; Kishima, M.; Nishimura, H.; Enomoto, S.; Hayashizaki, Y.; Suzuki, H. RUNX1 regulates site specificity of DNA demethylation by recruitment of DNA demethylation machineries in hematopoietic cells. Blood Adv. 2017, 1, 1699–1711. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Lin, H.-P.; Zhou, W.-J.; He, C.-X.; Zhang, Z.-Y.; Cheng, Z.-L.; Song, J.-B.; Liu, P.; Chen, X.-Y.; Xia, Y.-K.; et al. SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell Rep. 2018, 25, 1485–1500.e4. [Google Scholar] [CrossRef] [PubMed]
- Sardina, J.L.; Collombet, S.; Tian, T.V.; Gómez, A.; Di Stefano, B.; Berenguer, C.; Brumbaugh, J.; Stadhouders, R.; Segura-Morales, C.; Gut, M.; et al. Transcription Factors Drive Tet2-Mediated Enhancer Demethylation to Reprogram Cell Fate. Cell Stem. Cell 2018, 23, 905–906. [Google Scholar] [CrossRef]
- Chu, Y.; Zhao, Z.; Sant, D.W.; Zhu, G.; Greenblatt, S.M.; Liu, L.; Wang, J.; Cao, Z.; Tho, J.C.; Chen, S.; et al. Tet2 Regulates Osteoclast Differentiation by Interacting with Runx1 and Maintaining Genomic 5-Hydroxymethylcytosine (5hmC). Genom. Proteom. Bioinform. 2018, 16, 172–186. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef]
- Mondet, J.; Lo Presti, C.; Chevalier, S.; Bertrand, A.; Tondeur, S.; Blanchet, S.; Mc Leer, A.; Pernet-Gallay, K.; Mossuz, P. Mitochondria in human acute myeloid leukemia cell lines have ultrastructural alterations linked to deregulation of their respiratory profiles. Exp. Hematol. 2021, 98, 53–62.e3. [Google Scholar] [CrossRef]
- Chinnadurai, G.; Vijayalingam, S.; Rashmi, R. BIK, the founding member of the BH3-only family proteins: Mechanisms of cell death and role in cancer and pathogenic processes. Oncogene 2008, 27 (Suppl. S1), S20–S29. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Breeze, C.E.; Zheng, S.C.; Beck, S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinform. 2017, 18, 105. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, W.; Song, R.H.; Liu, S.; Wang, S.; Chen, Y.; Gao, C.; He, C.; Xiao, J.; Zhang, L.; et al. Tumor suppressor CEBPA interacts with and inhibits DNMT3A activity. Sci. Adv. 2022, 8, eabl5220. [Google Scholar] [CrossRef] [PubMed]
- Swart, L.E.; Heidenreich, O. The RUNX1/RUNX1T1 network: Translating insights into therapeutic options. Exp. Hematol. 2021, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Strissel, P.; Strick, R.; Chen, J.; Nucifora, G.; Le Beau, M.M.; Larson, R.A.; Rowley, J.D. Genomic DNA breakpoints in AML1/RUNX1 and ETO cluster with topoisomerase II DNA cleavage and DNase I hypersensitive sites in t(8;21) leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 3070–3075. [Google Scholar] [CrossRef] [PubMed]
- Tighe, J.E.; Calabi, F. t(8;21) breakpoints are clustered between alternatively spliced exons of MTG8. Clin. Sci. 1995, 89, 215–218. [Google Scholar] [CrossRef]
- Zampini, M.; Tregnago, C.; Bisio, V.; Simula, L.; Borella, G.; Manara, E.; Zanon, C.; Zonta, F.; Serafin, V.; Accordi, B.; et al. Epigenetic heterogeneity affects the risk of relapse in children with t(8;21)RUNX1-RUNX1T1-rearranged AML. Leukemia 2018, 32, 1124–1134. [Google Scholar] [CrossRef]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef]
- Lehnertz, B.; Zhang, Y.W.; Boivin, I.; Mayotte, N.; Tomellini, E.; Chagraoui, J.; Lavallée, V.-P.; Hébert, J.; Sauvageau, G. H3K27M/I mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 2017, 130, 2204–2214. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Heyn, H.; Vizoso, M.; Moutinho, C.; Vidal, E.; Gomez, A.; Cardus, A.M.; Simoriudalbas, L.; Moran, S.; Jost, E.; et al. DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene 2016, 35, 3079–3082. [Google Scholar] [CrossRef]
- Stahl, M.; Menghrajani, K.; Derkach, A.; Chan, A.; Xiao, W.; Glass, J.; King, A.C.; Daniyan, A.F.; Famulare, C.; Cuello, B.M.; et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021, 5, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68. [Google Scholar] [CrossRef] [PubMed]
- Rönnerblad, M.; Andersson, R.; Olofsson, T.; Douagi, I.; Karimi, M.; Lehmann, S.; Hoof, I.; De Hoon, M.; Itoh, M.; Nagao-Sato, S.; et al. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood 2014, 123, e79–e89. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ozark, P.A.; Smith, E.R.; Zhao, Z.; Marshall, S.A.; Rendleman, E.J.; Piunti, A.; Ryan, C.; Whelan, A.L.; Helmin, K.A.; et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Yevshin, I.; Sharipov, R.; Kolmykov, S.; Kondrakhin, Y.; Kolpakov, F. GTRD: A database on gene transcription regulation—2019 update. Nucleic Acids Res. 2019, 47, D100. [Google Scholar] [CrossRef]
- Lioznova, A.V.; Khamis, A.M.; Artemov, A.V.; Besedina, E.; Ramensky, V.; Bajic, V.B.; Kulakovskiy, I.V.; Medvedeva, Y.A. CpG traffic lights are markers of regulatory regions in human genome. BMC Genom. 2019, 20, 102. [Google Scholar] [CrossRef]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef]
- Qu, Y.; Lennartsson, A.; Gaidzik, V.I.; Deneberg, S.; Karimi, M.; Bengtzén, S.; Höglund, M.; Bullinger, L.; Döhner, K.; Lehmann, S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 2014, 9, 1108–1119. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Berest, I.; Keβler, S.; Nishimura, K.; Simón-Carrasco, L.; Vassiliou, G.S.; Pedersen, M.T.; Christensen, J.; Zaugg, J.B.; Helin, K. TET2 binding to enhancers facilitates transcription factor recruitment in hematopoietic cells. Genome Res. 2019, 29, 564–575. [Google Scholar] [CrossRef] [Green Version]
- “MONOMAC1.” n.d. Available online: https://depmap.org/portal/cell_line/ACH-001129?tab=mutation (accessed on 12 April 2021).
- The Institute for Cancer Research, UK. n.d. “canSAR Black.” The Institute for Cancer Research, UK. Available online: https://cansarblack.icr.ac.uk/cell-line/OCI-AML-5/mutations (accessed on 12 April 2021).


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanova, E.I.; Zubritskiy, A.V.; Lioznova, A.V.; Ogunleye, A.J.; Golotin, V.A.; Guts, A.A.; Lennartsson, A.; Demidov, O.N.; Medvedeva, Y.A. RUNX1/CEBPA Mutation in Acute Myeloid Leukemia Promotes Hypermethylation and Indicates for Demethylation Therapy. Int. J. Mol. Sci. 2022, 23, 11413. https://doi.org/10.3390/ijms231911413
Romanova EI, Zubritskiy AV, Lioznova AV, Ogunleye AJ, Golotin VA, Guts AA, Lennartsson A, Demidov ON, Medvedeva YA. RUNX1/CEBPA Mutation in Acute Myeloid Leukemia Promotes Hypermethylation and Indicates for Demethylation Therapy. International Journal of Molecular Sciences. 2022; 23(19):11413. https://doi.org/10.3390/ijms231911413
Chicago/Turabian StyleRomanova, Ekaterina I., Anatoliy V. Zubritskiy, Anna V. Lioznova, Adewale J. Ogunleye, Vasily A. Golotin, Anna A. Guts, Andreas Lennartsson, Oleg N. Demidov, and Yulia A. Medvedeva. 2022. "RUNX1/CEBPA Mutation in Acute Myeloid Leukemia Promotes Hypermethylation and Indicates for Demethylation Therapy" International Journal of Molecular Sciences 23, no. 19: 11413. https://doi.org/10.3390/ijms231911413
APA StyleRomanova, E. I., Zubritskiy, A. V., Lioznova, A. V., Ogunleye, A. J., Golotin, V. A., Guts, A. A., Lennartsson, A., Demidov, O. N., & Medvedeva, Y. A. (2022). RUNX1/CEBPA Mutation in Acute Myeloid Leukemia Promotes Hypermethylation and Indicates for Demethylation Therapy. International Journal of Molecular Sciences, 23(19), 11413. https://doi.org/10.3390/ijms231911413