

CRISPR/Cas9—A Promising Therapeutic Tool to Cure Blindness: Current Scenario and Future Prospects


Abstract
:1. Introduction
2. Genetic Causes of Retinal Degeneration and Blindness
3. Classical Therapeutic Approaches to Restore the Vision
4. CRISPR/Cas9 Is a Promising Strategy to Restore the Blindness
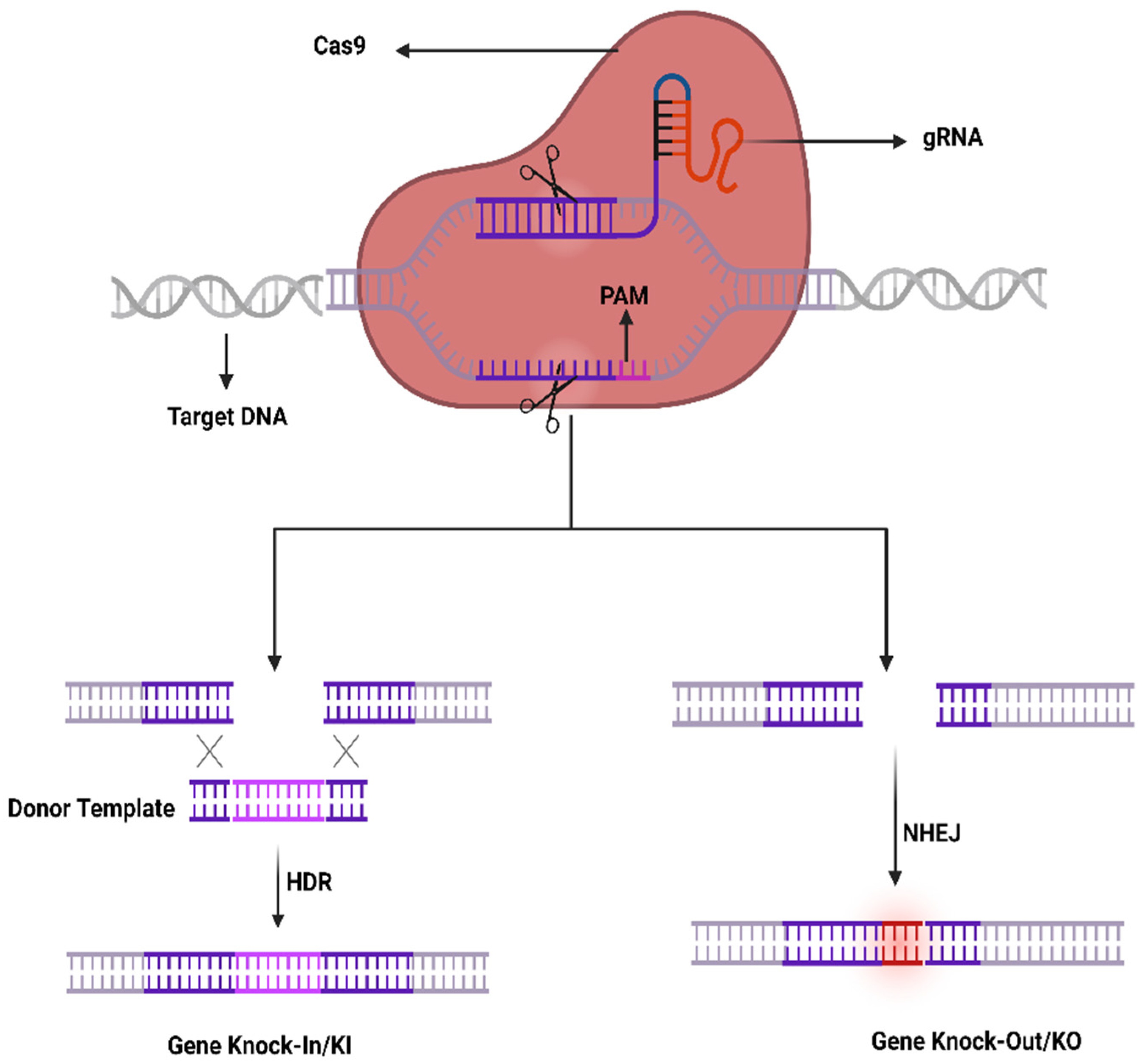
4.1. CRISPR-Cas System; Development, Components, and Mechanism
4.2. CRISPR-Cas System Components Delivery to Ocular Tissues
4.2.1. CRISPR to Treat AMD
4.2.2. CRISPR to Treat the Glaucoma
4.2.3. CRISPR to Treat Retinitis Pigmentosa (RP)
4.2.4. CRISPR to Treat Leber Congenital Amaurosis (LCA10)
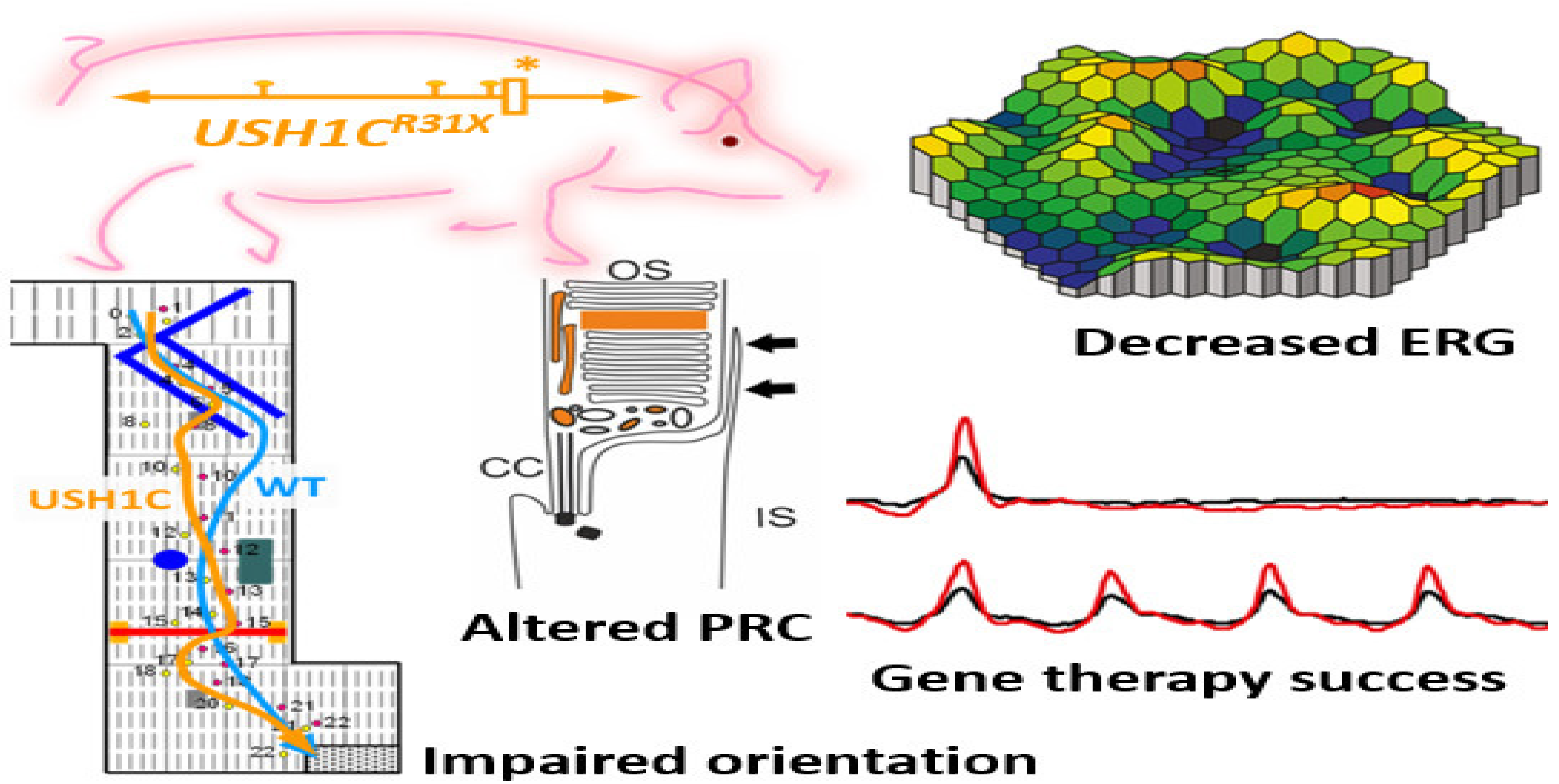
4.2.5. CRISPR to Treat the Usher Syndrome
5. Base and Prime Editing: Advanced Gene Editing Tools to Cure Blindness
6. Limitations of CRISPR/Cas9 in Clinical Applications to Cure the Blindness
6.1. Delivery
6.2. Specificity or Precision Editing
6.3. Off-Target Effects
7. Conclusions and Future Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flaxman, S.R.; Bourne, R.R.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Amitai, G.; Sorek, R. CRISPR–Cas Adaptation: Insights into the Mechanism of Action. Nat. Rev. Microbiol. 2016, 14, 67–76. [Google Scholar] [CrossRef]
- Single Ascending Dose Study in Participants with LCA10. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03872479 (accessed on 15 July 2022).
- Safety and Efficacy of CRISPR/Cas9 mRNA Instantaneous Gene Editing Therapy to Treat Refractory Viral Keratitis. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04560790 (accessed on 24 August 2022).
- Ruan, G.; Barry, E.; Lukason, M.; Cheng, S.; Scaria, A. Using CRISPR/Cas9 as a Therapeutic Approach for Leber Congenital Amaurosis 10 (LCA10). Mol. Ther. 2016, 24, S131–S132. [Google Scholar] [CrossRef]
- Tan, P.L.; Bowes Rickman, C.; Katsanis, N. AMD and the alternative complement pathway: Genetics and functional implications. Hum. Genom. 2016, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Tyagi, S.C. Genes and genetics in eye diseases: A genomic medicine approach for investigating hereditary and inflammatory ocular disorders. Int. J. Ophthalmol. 2018, 11, 117. [Google Scholar] [PubMed]
- Liu, M.M.; Chan, C.-C.; Tuo, J. Genetic mechanisms and age-related macular degeneration: Common variants, rare variants, copy number variations, epigenetics, and mitochondrial genetics. Hum. Genom. 2012, 6, 13. [Google Scholar] [CrossRef]
- Ayala-Haedo, J.A.; Gallins, P.J.; Whitehead, P.L.; Schwartz, S.G.; Kovach, J.L.; Postel, E.A.; Agarwal, A.; Wang, G.; Haines, J.L.; Pericak-Vance, M.A. Analysis of Single Nucleotide Polymorphisms in the NOS2A Gene and Interaction with Smoking in Age-Related Macular Degeneration. Ann. Hum. Genet. 2010, 74, 195–201. [Google Scholar] [CrossRef]
- Sohn, E.H.; Han, I.C.; Roos, B.R.; Faga, B.; Luse, M.A.; Binkley, E.M.; Boldt, H.C.; Folk, J.C.; Russell, S.R.; Mullins, R.F.; et al. Genetic Association between MMP9 and Choroidal Neovascularization in Age-Related Macular Degeneration. Ophthalmol. Sci. 2021, 1, 100002. [Google Scholar] [CrossRef]
- Dewing, J.M.; Carare, R.O.; Lotery, A.J.; Ratnayaka, J.A. The Diverse Roles of TIMP-3: Insights into Degenerative Diseases of the Senescent Retina and Brain. Cells 2020, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.H.; Bell, B.; Singh, R.; Batoki, J.; Wolk, A.; Cutler, A.; Prayson, N.; Ali, M.; Stoehr, H.; Anand-Apte, B. Sorsby Fundus Dystrophy Mutation in Tissue Inhibitor of Metalloproteinase 3 (TIMP3) promotes Choroidal Neovascularization via a Fibroblast Growth Factor-dependent Mechanism. Sci. Rep. 2019, 9, 17429. [Google Scholar] [CrossRef] [PubMed]
- Liutkeviciene, R.; Vilkeviciute, A.; Gedvilaite, G.; Kaikaryte, K.; Kriauciuniene, L. Haplotypes of HTRA1 rs1120638, TIMP3 rs9621532, VEGFA rs833068, CFI rs10033900, ERCC6 rs3793784, and KCTD10 rs56209061 Gene Polymorphisms in Age-Related Macular Degeneration. Dis. Markers 2019, 2019, 9602949. [Google Scholar] [CrossRef] [PubMed]
- Vishal, M.; Sharma, A.; Kaurani, L.; Alfano, G.; Mookherjee, S.; Narta, K.; Agrawal, J.; Bhattacharya, I.; Roychoudhury, S.; Ray, J. Genetic association and stress mediated down-regulation in trabecular meshwork implicates MPP7 as a novel candidate gene in primary open angle glaucoma. BMC Med. Genom. 2016, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Thorleifsson, G.; Walters, G.B.; Hewitt, A.W.; Masson, G.; Helgason, A.; DeWan, A.; Sigurdsson, A.; Jonasdottir, A.; Gudjonsson, S.A.; Magnusson, K.P. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat. Genet. 2010, 42, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Pasutto, F.; Krumbiegel, M.; Mardin, C.Y.; Paoli, D.; Lämmer, R.; Weber, B.H.; Kruse, F.E.; Schlötzer-Schrehardt, U.; Reis, A. Association of LOXL1 common sequence variants in German and Italian patients with pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1459–1463. [Google Scholar] [CrossRef]
- Patel, N.; Anand, D.; Monies, D.; Maddirevula, S.; Khan, A.O.; Algoufi, T.; Alowain, M.; Faqeih, E.; Alshammari, M.; Qudair, A. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum. Genet. 2017, 136, 205–225. [Google Scholar] [CrossRef]
- Ahmad, R.; Bell, S.; Moosajee, M. Genetics of Congenital Cataract. Adv. Ophthalmol. Optom. 2022, 7, 89–118. [Google Scholar] [CrossRef]
- Shentu, X.; Miao, Q.; Tang, X.; Yin, H.; Zhao, Y. Identification and functional analysis of a novel MIP gene mutation associated with congenital cataract in a Chinese family. PLoS ONE 2015, 10, e0126679. [Google Scholar] [CrossRef]
- Qin, L.; Guo, L.; Wang, H.; Li, T.; Lou, G.; Guo, Q.; Hou, Q.; Liu, H.; Liao, S.; Liu, Z. A novel MIP mutation in familial congenital nuclear cataracts. Eur. J. Med. Genet. 2016, 59, 488–491. [Google Scholar] [CrossRef]
- Yanovitch, T.; Li, Y.-J.; Metlapally, R.; Abbott, D.; Viet, K.-N.T.; Young, T.L. Hepatocyte growth factor and myopia: Genetic association analyses in a Caucasian population. Mol. Vis. 2009, 15, 1028. [Google Scholar]
- Nishizaki, R.; Ota, M.; Inoko, H.; Meguro, A.; Shiota, T.; Okada, E.; Mok, J.; Oka, A.; Ohno, S.; Mizuki, N. New susceptibility locus for high myopia is linked to the uromodulin-like 1 (UMODL1) gene region on chromosome 21q22. 3. Eye 2009, 23, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Xie, Y.; Zernant, J.; Yuan, B.; Bearelly, S.; Tsang, S.H.; Lupski, J.R.; Allikmets, R. Complex inheritance of ABCA4 disease: Four mutations in a family with multiple macular phenotypes. Hum. Genet. 2016, 135, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, F.; Burstedt, M.S.; Sandgren, O.; Norberg, A.; Golovleva, I. Novel mutations in CRB1 and ABCA4 genes cause Leber congenital amaurosis and Stargardt disease in a Swedish family. Eur. J. Hum. Genet. 2013, 21, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhou, X.; Wang, F.; Yan, M.; Ding, F. Evidence for a novel autosomal dominant retinitis pigmentosa linked to chromosome 1p22. 1-q12 in a Chinese family. Curr. Eye Res. 2011, 36, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hoyos, M.; Garcia-Sandoval, B.; Cantalapiedra, D.; Riveiro, R.; Lorda-Sánchez, I.; Trujillo-Tiebas, M.J.; de Alba, M.R.; Millan, J.M.; Baiget, M.; Ramos, C. Mutational screening of the RP2 and RPGR genes in Spanish families with X-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3777–3782. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.S.; Bowne, S.J.; Seaman, C.R.; Blanton, S.H.; Lewis, R.A.; Heckenlively, J.R.; Birch, D.G.; Hughbanks-Wheaton, D.; Daiger, S.P. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4579–4588. [Google Scholar] [CrossRef]
- Chizzolini, M.; Galan, A.; Milan, E.; Sebastiani, A.; Costagliola, C.; Parmeggiani, F. Good epidemiologic practice in retinitis pigmentosa: From phenotyping to biobanking. Curr. Genom. 2011, 12, 260–266. [Google Scholar]
- Al-Maskari, A.; O’grady, A.; Pal, B.; McKibbin, M. Phenotypic progression in X-linked retinitis pigmentosa secondary to a novel mutation in the RPGR gene. Eye 2009, 23, 519–521. [Google Scholar] [CrossRef]
- Branham, K.; Matsui, H.; Biswas, P.; Guru, A.A.; Hicks, M.; Suk, J.J.; Li, H.; Jakubosky, D.; Long, T.; Telenti, A. Establishing the involvement of the novel gene AGBL5 in retinitis pigmentosa by whole genome sequencing. Physiol. Genom. 2016, 48, 922–927. [Google Scholar] [CrossRef]
- Faivre, L.; Collod-Beroud, G.; Callewaert, B.; Child, A.; Binquet, C.; Gautier, E.; Loeys, B.L.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M. Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24–32 mutation. Eur. J. Hum. Genet. 2009, 17, 491–501. [Google Scholar] [CrossRef]
- Faivre, L.; Collod-Beroud, G.; Callewaert, B.; Child, A.; Loeys, B.; Binquet, C.; Gautier, E.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M. Pathogenic FBN1 mutations in 146 adults not meeting clinical diagnostic criteria for Marfan syndrome: Further delineation of type 1 fibrillinopathies and focus on patients with an isolated major criterion. Am. J. Med. Genet. Part A 2009, 149, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Masurel-Paulet, A.; Collod-Beroud, G.; Callewaert, B.L.; Child, A.H.; Stheneur, C.; Binquet, C.; Gautier, E.; Chevallier, B.; Huet, F. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics 2009, 123, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lai, Y.H.; Capasso, J.E.; Han, S.; Levin, A.V. Early onset ectopia lentis due to a FBN1 mutation with non-penetrance. Am. J. Med. Genet. Part A 2015, 167, 1365–1368. [Google Scholar] [CrossRef]
- Choi, Y.M.; Shim, K.S.; Yoon, K.L.; Han, M.Y.; Cha, S.H.; Kim, S.K.; Jung, J.H. Transforming growth factor beta receptor II polymorphisms are associated with Kawasaki disease. Korean J. Pediatrics 2012, 55, 18. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nakayama, T.; Mori, R.; Sato, N.; Kawamura, A.; Yuzawa, M. Associations of complement factor B and complement component 2 genotypes with subtypes of polypoidal choroidal vasculopathy. BMC Ophthalmol. 2014, 14, 83. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lai, T.Y.; Chiang, S.W.; Chan, V.C.; Young, A.L.; Tam, P.O.; Pang, C.P.; Chen, L.J. Gender specific association of a complement component 3 polymorphism with polypoidal choroidal vasculopathy. Sci. Rep. 2014, 4, 7018. [Google Scholar] [CrossRef]
- Liu, K.; Lai, T.Y.; Ma, L.; Lai, F.H.; Young, A.L.; Brelen, M.E.; Tam, P.O.; Pang, C.P.; Chen, L.J. Ethnic differences in the association of SERPING1 with age-related macular degeneration and polypoidal choroidal vasculopathy. Sci. Rep. 2015, 5, 9424. [Google Scholar] [CrossRef]
- Metz, C.; Lohmann, D.; Zeschnigk, M.; Bornfeld, N. Uveal melanoma: Current insights into clinical relevance of genetic testing. Klin. Mon. Fur Augenheilkd. 2013, 230, 686–691. [Google Scholar]
- Werdich, X.Q.; Jakobiec, F.A.; Singh, A.D.; Kim, I.K. A review of advanced genetic testing for clinical prognostication in uveal melanoma. Semin. Ophthalmol. 2013, 28, 361–371. [Google Scholar] [CrossRef]
- Buecher, B.; Gauthier-Villars, M.; Desjardins, L.; Rouic, L.-L.; Levy, C.; De Pauw, A.; Bombled, J.; Tirapo, C.; Houdayer, C.; Bressac-de Paillerets, B. Contribution of CDKN2A/P16 INK4A, P14 ARF, CDK4 and BRCA1/2 germline mutations in individuals with suspected genetic predisposition to uveal melanoma. Fam. Cancer 2010, 9, 663–667. [Google Scholar] [CrossRef]
- Cohn, A.C.; Toomes, C.; Hewitt, A.W.; Kearns, L.S.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. The natural history of OPA1-related autosomal dominant optic atrophy. Br. J. Ophthalmol. 2008, 92, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Toomes, C.; Marchbank, N.J.; Mackey, D.A.; Craig, J.E.; Newbury-Ecob, R.A.; Bennett, C.P.; Vize, C.J.; Desai, S.P.; Black, G.C.; Patel, N. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum. Mol. Genet. 2001, 10, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Khanna, H.; Punzo, C. Advances in gene therapy for diseases of the eye. Hum. Gene Ther. 2016, 27, 563–579. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Weiland, J.D.; Roska, B.; Humayun, M.S. Retinal stimulation strategies to restore vision: Fundamentals and systems. Prog. Retin. Eye Res. 2016, 53, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Ayton, L.N.; Barnes, N.; Dagnelie, G.; Fujikado, T.; Goetz, G.; Hornig, R.; Jones, B.W.; Muqit, M.K.; Rathbun, D.L.; Stingl, K.; et al. An update on retinal prostheses. Clin. Neurophysiol. 2020, 131, 1383–1398. [Google Scholar] [CrossRef]
- Brown, B. FDA Approves Upgraded System for the Blind. 2021. Available online: https://healthtechinsider.com/2021/03/08/fda-approves-upgraded-system-for-the-blind/ (accessed on 8 March 2021).
- Nanegrungsunk, O.; Au, A.; Sarraf, D.; Sadda, S.R. New frontiers of retinal therapeutic intervention: A critical analysis of novel approaches. Ann. Med. 2022, 54, 1067–1080. [Google Scholar] [CrossRef]
- Beauchamp, M.S.; Oswalt, D.; Sun, P.; Foster, B.L.; Magnotti, J.F.; Niketeghad, S.; Pouratian, N.; Bosking, W.H.; Yoshor, D. Dynamic Stimulation of Visual Cortex Produces Form Vision in Sighted and Blind Humans. Cell 2020, 181, 774–783. [Google Scholar] [CrossRef]
- Scholl, H.P.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.-A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, rv366–rv368. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V. Treatment possibilities for retinitis pigmentosa. N. Engl. J. Med. 2010, 363, 1669–1671. [Google Scholar] [CrossRef]
- Pardue, M.T.; Allen, R.S. Neuroprotective strategies for retinal disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef]
- Wubben, T.J.; Besirli, C.G.; Johnson, M.W.; Zacks, D.N. Retinal neuroprotection: Overcoming the translational roadblocks. Am. J. Ophthalmol. 2018, 192, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.C.; Humayun, M.S.; Dorn, J.D.; Da Cruz, L.; Dagnelie, G.; Handa, J.; Barale, P.-O.; Sahel, J.-A.; Stanga, P.E.; Hafezi, F. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology 2015, 122, 1547–1554. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Roska, B. Gene therapy for blindness. Annu. Rev. Neurosci. 2013, 36, 467–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Goldman, D. Retina regeneration in zebrafish. Curr. Opin. Genet. Dev. 2016, 40, 41–47. [Google Scholar] [CrossRef] [PubMed]
- da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M. Phase 1 clinical study of an embryonic stem cell–derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.C.; Hallam, D.; Karimi, A.; Mellough, C.B.; Chen, J.; Steel, D.H.; Lako, M. 3D culture of human pluripotent stem cells in RGD-alginate hydrogel improves retinal tissue development. Acta Biomater. 2017, 49, 329–343. [Google Scholar] [CrossRef]
- Lamba, D.A.; Gust, J.; Reh, T.A. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell 2009, 4, 73–79. [Google Scholar] [CrossRef]
- Jansen, R.; Embden, J.D.v.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef]
- Kunin, V.; Sorek, R.; Hugenholtz, P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007, 8, R61. [Google Scholar] [CrossRef]
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cas9, Cpf1 and C2c1/2/3—What’s next? Bioengineered 2017, 8, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Mollhoff, I.N.; Nguyen, U.; Nguyen, A.; Stucka, N.; Tieu, E.; Manna, S.; Meleppat, R.K.; Zhang, P.; Nguyen, E.L. Factors impacting efficacy of AAV-mediated CRISPR-based genome editing for treatment of choroidal neovascularization. Mol. Ther.-Methods Clin. Dev. 2020, 17, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef]
- Kim, J.; Hwang, J.-M.; Ko, H.; Seong, M.-W.; Park, B.-J.; Park, S. Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 2005, 64, 966–972. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Efficient delivery of genome-editing proteins in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73. [Google Scholar] [CrossRef]
- Kuo, J.Z.; Wong, T.Y.; Ong, F.S. Genetic risk, ethnic variations and pharmacogenetic biomarkers in AMD and polypoidal choroidal vasculopathy. Expert Rev. Ophthalmol. 2013, 8, 127–140. [Google Scholar] [CrossRef]
- Imamura, Y.; Engelbert, M.; Iida, T.; Freund, K.B.; Yannuzzi, L.A. Polypoidal choroidal vasculopathy: A review. Surv. Ophthalmol. 2010, 55, 501–515. [Google Scholar] [CrossRef]
- Kim, K.; Park, S.W.; Kim, J.H.; Lee, S.H.; Kim, D.; Koo, T.; Kim, K.-e.; Kim, J.H.; Kim, J.-S. Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res. 2017, 27, 419–426. [Google Scholar] [CrossRef]
- Ling, S.; Yang, S.; Hu, X.; Yin, D.; Dai, Y.; Qian, X.; Wang, D.; Pan, X.; Hong, J.; Sun, X. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 2021, 5, 144–156. [Google Scholar] [CrossRef]
- Ma, M.; Li, Z.; Wang, D.W.; Wei, X. Next-generation sequencing identifies novel mutations in the FBN1 gene for two Chinese families with Marfan syndrome. Mol. Med. Rep. 2016, 14, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Laude, A.; Cackett, P.D.; Vithana, E.N.; Yeo, I.Y.; Wong, D.; Koh, A.H.; Wong, T.Y.; Aung, T. Polypoidal choroidal vasculopathy and neovascular age-related macular degeneration: Same or different disease? Prog. Retin. Eye Res. 2010, 29, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Zode, G.; Kasetti, R.B.; Ran, F.A.; Yan, W.; Sharma, T.P.; Bugge, K.; Searby, C.C.; Fingert, J.H.; Zhang, F. CRISPR-Cas9–based treatment of myocilin-associated glaucoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayana, N.P.; Sugali, C.K.; Dai, J.; Peng, M.; Liu, S.; Zhang, Y.; Wan, J.; Mao, W. Using CRISPR Interference as a Therapeutic Approach to Treat TGFβ2-Induced Ocular Hypertension and Glaucoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 7. [Google Scholar] [CrossRef]
- Wu, J.; Bell, O.H.; Copland, D.A.; Young, A.; Pooley, J.R.; Maswood, R.; Evans, R.S.; Khaw, P.T.; Ali, R.R.; Dick, A.D. Gene therapy for glaucoma by ciliary body aquaporin 1 disruption using CRISPR-Cas9. Mol. Ther. 2020, 28, 820–829. [Google Scholar] [CrossRef]
- Coupland, S.E.; Lake, S.L.; Zeschnigk, M.; Damato, B. Molecular pathology of uveal melanoma. Eye 2013, 27, 230–242. [Google Scholar] [CrossRef]
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol. Ther. 2016, 24, 556–563. [Google Scholar] [CrossRef]
- Xu, C.L.; Park, K.S.; Tsang, S.H. CRISPR/Cas9 genome surgery for retinal diseases. Drug Discov. Today Technol. 2018, 28, 23–32. [Google Scholar] [CrossRef]
- Shahin, S.; Xu, H.; Lu, B.; Mercado, A.; Jones, M.K.; Bakondi, B.; Wang, S. AAV-CRISPR/Cas9 Gene Editing Preserves Long-Term Vision in the P23H Rat Model of Autosomal Dominant Retinitis Pigmentosa. Pharmaceutics 2022, 14, 824. [Google Scholar] [CrossRef]
- Gumerson, J.D.; Alsufyani, A.; Yu, W.; Lei, J.; Sun, X.; Dong, L.; Wu, Z.; Li, T. Restoration of RPGR expression in vivo using CRISPR/Cas9 gene editing. Gene Ther. 2022, 29, 81–93. [Google Scholar] [CrossRef]
- Grotz, S.; Schäfer, J.; Wunderlich, K.A.; Ellederova, Z.; Auch, H.; Bähr, A.; Runa-Vochozkova, P.; Fadl, J.; Arnold, V.; Ardan, T. Early disruption of photoreceptor cell architecture and loss of vision in a humanized pig model of usher syndromes. EMBO Mol. Med. 2022, 14, e14817. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest developed strategies to minimize the off-target effects in CRISPR-Cas-mediated genome editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, P.; Miccio, A.; Brusson, M. Base and prime editing technologies for blood disorders. Front. Genome Ed. 2021, 3, 618406. [Google Scholar] [CrossRef] [PubMed]
- Sakata, R.C.; Ishiguro, S.; Mori, H.; Tanaka, M.; Tatsuno, K.; Ueda, H.; Yamamoto, S.; Seki, M.; Masuyama, N.; Nishida, K. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol. 2020, 38, 865–869. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Choi, E.H.; Suh, S.; Foik, A.T.; Leinonen, H.; Newby, G.A.; Gao, X.D.; Banskota, S.; Hoang, T.; Du, S.W.; Dong, Z. In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration. Nat. Commun. 2022, 13, 1830. [Google Scholar] [CrossRef]
- Zhi, S.; Chen, Y.; Wu, G.; Wen, J.; Wu, J.; Liu, Q.; Li, Y.; Kang, R.; Hu, S.; Wang, J. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2022, 30, 283–294. [Google Scholar] [CrossRef]
- Höijer, I.; Emmanouilidou, A.; Östlund, R.; van Schendel, R.; Bozorgpana, S.; Tijsterman, M.; Feuk, L.; Gyllensten, U.; den Hoed, M.; Ameur, A. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun. 2022, 13, 627. [Google Scholar] [CrossRef]
- Artegiani, B.; Hendriks, D.; Beumer, J.; Kok, R.; Zheng, X.; Joore, I.; Chuva de Sousa Lopes, S.; van Zon, J.; Tans, S.; Clevers, H. Fast and efficient generation of knock-in human organoids using homology-independent CRISPR–Cas9 precision genome editing. Nat. Cell Biol. 2020, 22, 321–331. [Google Scholar] [CrossRef]
- Muruve, D.A. The innate immune response to adenovirus vectors. Hum. Gene Ther. 2004, 15, 1157–1166. [Google Scholar] [CrossRef]
- Nakai, H.; Yant, S.R.; Storm, T.A.; Fuess, S.; Meuse, L.; Kay, M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001, 75, 6969–6976. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, F.; Gao, G. CRISPR-based therapeutic genome editing: Strategies and in vivo delivery by AAV vectors. Cell 2020, 181, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Ouyang, K.; Xu, X.; Xu, L.; Wen, C.; Zhou, X.; Qin, Z.; Xu, Z.; Sun, W.; Liang, Y. Nanoparticle delivery of CRISPR/Cas9 for genome editing. Front. Genet. 2021, 12, 673286. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Hoque, M.Z.; Ovais, M.; Basheer, C.; Ahmad, I. Stimulus-Responsive Smart Nanoparticles-Based CRISPR-Cas Delivery for Therapeutic Genome Editing. Int. J. Mol. Sci. 2021, 22, 11300. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Zhang, X.; Quan, R.; Zhong, C.; Mo, J.; Huang, Y.; Wang, H.; Ruan, X.; Xu, Z. Suppressing Ku70/Ku80 expression elevates homology-directed repair efficiency in primary fibroblasts. Int. J. Biochem. Cell Biol. 2018, 99, 154–160. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Welker, J.M.; Wierson, W.A.; Almeida, M.P.; Mann, C.M.; Torrie, M.E.; Ming, Z.; Ekker, S.C.; Clark, K.J.; Dobbs, D.L.; Essner, J.J. GeneWeld: Efficient Targeted Integration Directed by Short Homology in Zebrafish. Bio-Protoc. 2021, 11, e4100. [Google Scholar] [CrossRef]
- Nishiyama, J.; Mikuni, T.; Yasuda, R. Virus-mediated genome editing via homology-directed repair in mitotic and postmitotic cells in mammalian brain. Neuron 2017, 96, 755–768.e755. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther.-Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef]
- Aquino-Jarquin, G. Current advances in overcoming obstacles of CRISPR/Cas9 off-target genome editing. Mol. Genet. Metab. 2021, 134, 77–86. [Google Scholar] [CrossRef]
- Ledford, H. CRISPR treatment inserted directly into the body for first time. Nature 2020, 579, 185–186. [Google Scholar] [CrossRef]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Eye Disease (Onset Age) | Gene Variants |
|---|---|---|
| 1 | AMD (50–60 years) | Nitric oxide synthase 2A (NOS2A), tissue inhibitor of matrix metalloproteinase 3 (TIMP-3), matrix metalloproteinase-9 (MMP-9), high-temperature requirement factor A serine peptidase 1 (HTRA1), excision repair cross-complementing group 6 (ERCC6), etc. |
| 2 | Glaucoma (>40 excluding congenital form in infants) | Mouse myocilin (MYOC), paired-like homeodomain2 (PITX2), paired box protein (PAX6), cytochrome p450-1B1 (CYP1B1), latent TGF-β binding protein-2 (LTBP2), etc. |
| 3 | Cataract (50–60 years) | Transmembrane anterior posterior transformation 1 (TAPT1), gem nuclear organelle associated protein 4 (GEMIN4), AP endonuclease 1 (APE1), major intrinsic protein (MIP), etc. |
| 4 | Myopia (progresses in ~ age 20) | Hepatocyte growth factor (HGF), mesenchymal-epithelial transition factor (C-MET), uromodulin like 1 (UMODL1), paired box protein (PAX6), etc. |
| 5 | Stargardt’s disease (early childhood to middle age) | ATP binding cassette subfamily A member 4 (ABCA4), crumbs homology 1 (CRB1), etc. |
| 6 | Retinitis pigmentosa (10–30 years) | Retinitis pigmentosa GTPase regulator (RPGR), precursor mRNA processing factor 3 (PRPF3), ATP/GTP binding like 5 (AGBL5), etc. |
| 7 | Marfan syndrome (newborn babies that may be later on) | fibrillin 1 (FBN1), transforming growth factor beta receptor 2 (TGFBR2), etc. |
| 8 | Polypoidal choroidal vasculopathies (55–65 years) | Complement 2 (C2), complement 3 (C3), complement factor B (CFB), erpin peptidase inhibitor clade G member 1 (SERPING1), pigment epithelium-derived factor (PEDF), etc. |
| 9 | Uveal melanoma (50–80 years) | Ubiquitin carboxyl-terminal hydrolase (BAP1), guanine nucleotide-binding protein G(q) (GNAQ), G protein subunit alpha 11 (GNA11), spliceosome factor 3B subunit 1 (SF3B1), eukaryotic translation initiation factor 1A X-chromosome (EIF1AX), etc. |
| 10 | Inherited optic neuropathies (X-linked appears in young males) | Complex I or ND genes, OPA1, RPE65, etc. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, I. CRISPR/Cas9—A Promising Therapeutic Tool to Cure Blindness: Current Scenario and Future Prospects. Int. J. Mol. Sci. 2022, 23, 11482. https://doi.org/10.3390/ijms231911482
Ahmad I. CRISPR/Cas9—A Promising Therapeutic Tool to Cure Blindness: Current Scenario and Future Prospects. International Journal of Molecular Sciences. 2022; 23(19):11482. https://doi.org/10.3390/ijms231911482
Chicago/Turabian StyleAhmad, Irshad. 2022. "CRISPR/Cas9—A Promising Therapeutic Tool to Cure Blindness: Current Scenario and Future Prospects" International Journal of Molecular Sciences 23, no. 19: 11482. https://doi.org/10.3390/ijms231911482
APA StyleAhmad, I. (2022). CRISPR/Cas9—A Promising Therapeutic Tool to Cure Blindness: Current Scenario and Future Prospects. International Journal of Molecular Sciences, 23(19), 11482. https://doi.org/10.3390/ijms231911482