


PLA2G2A Phospholipase Promotes Fatty Acid Synthesis and Energy Metabolism in Pancreatic Cancer Cells with K-ras Mutation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
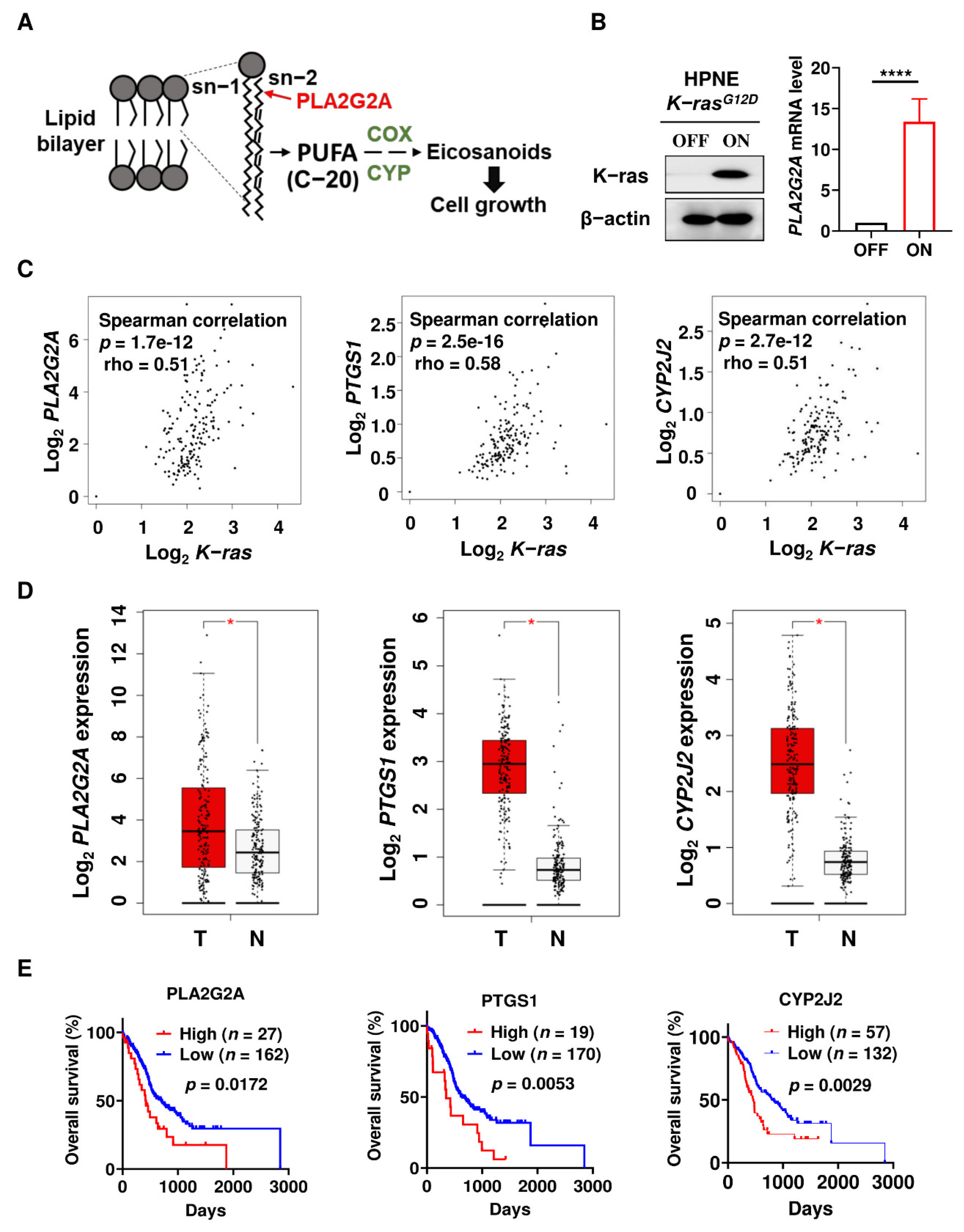
2.1. Activation of Mutant K-rasG12D Induces PLA2G2A Expression Which Is Correlated with Poor Prognosis in PDAC Patients
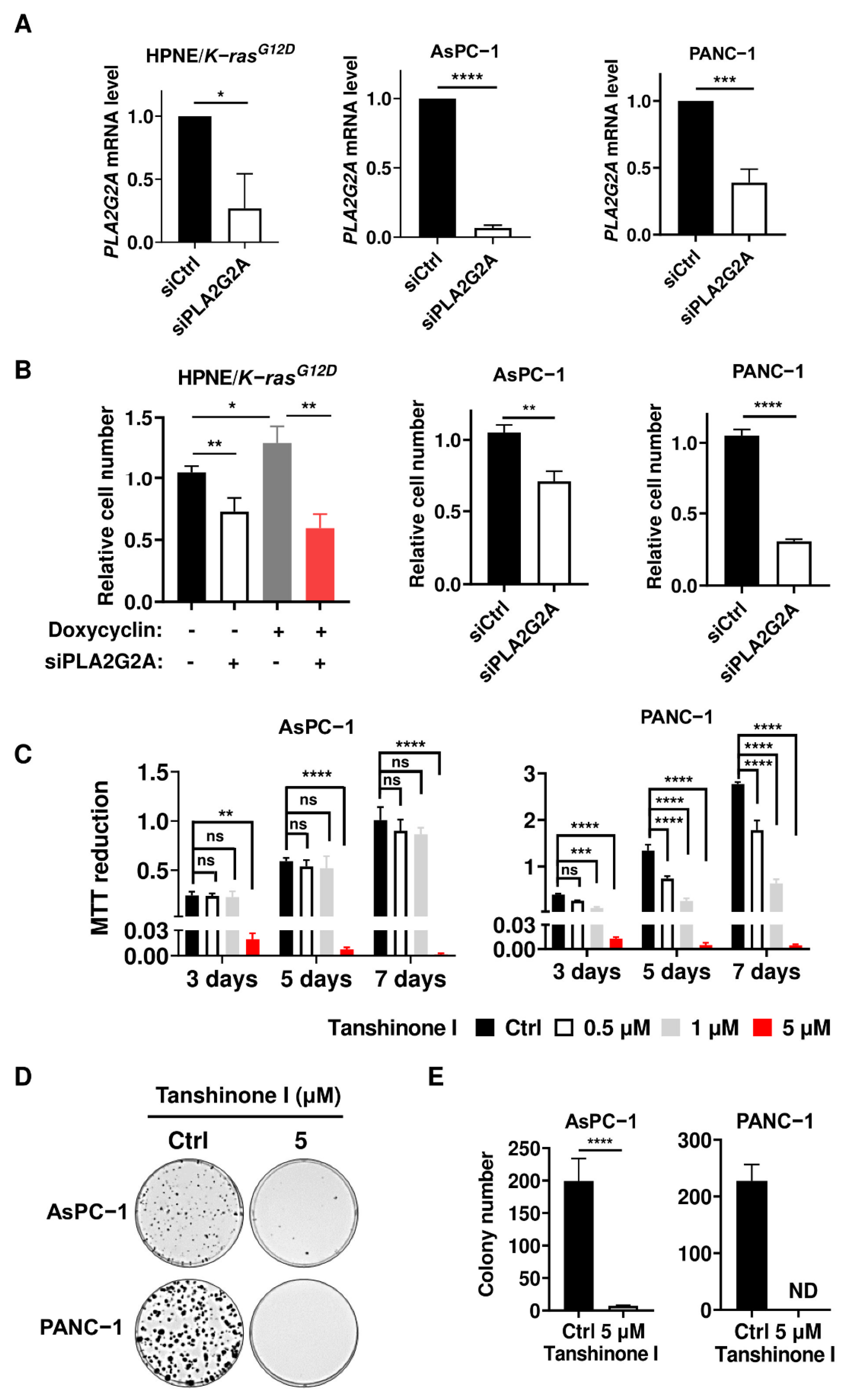
2.2. Abrogation of PLA2G2A Suppresses PDAC Cell Proliferation
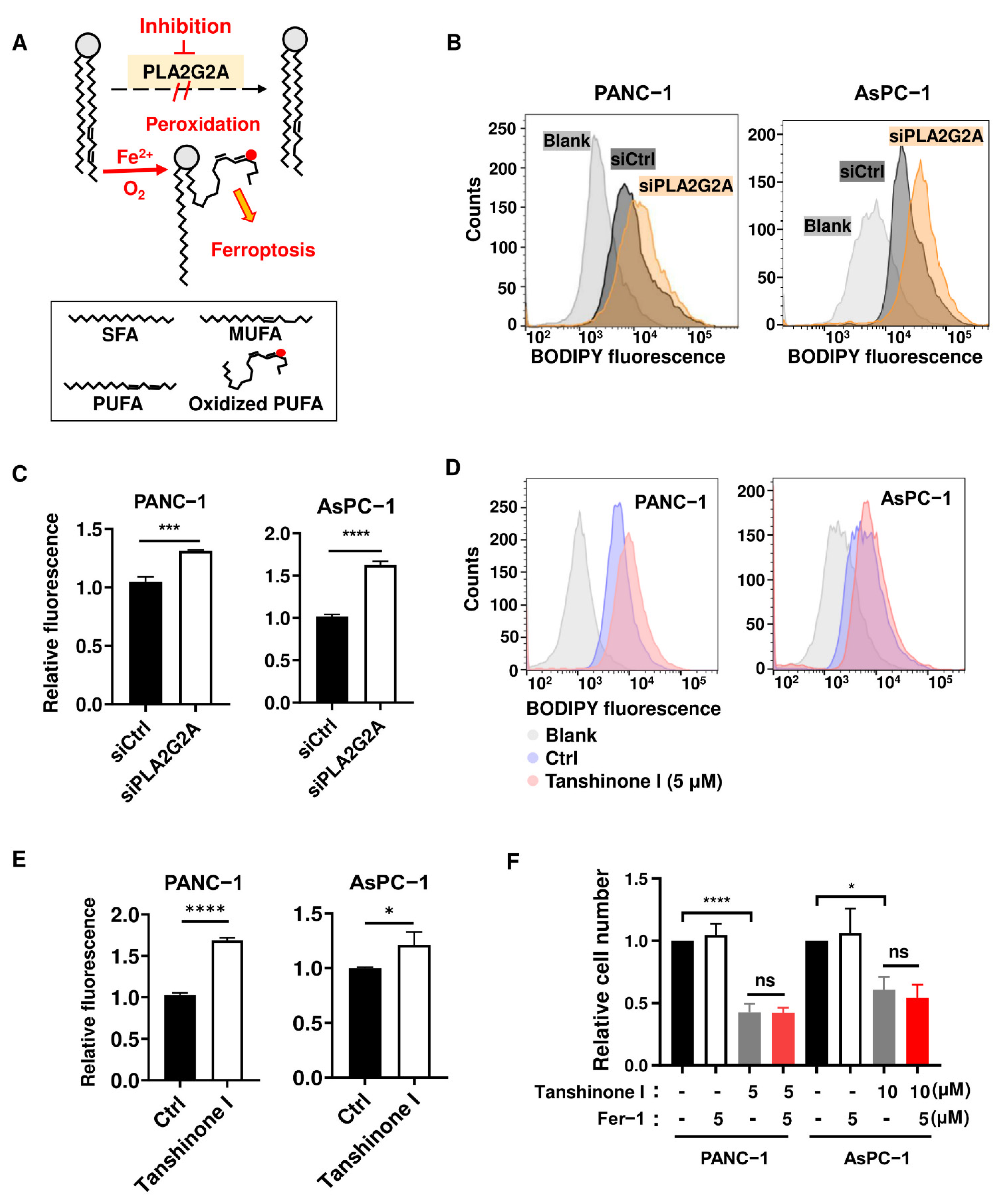
2.3. Abrogation of PLA2G2A Induces a Moderate Increase in Lipid Peroxidation without Causing Ferroptosis
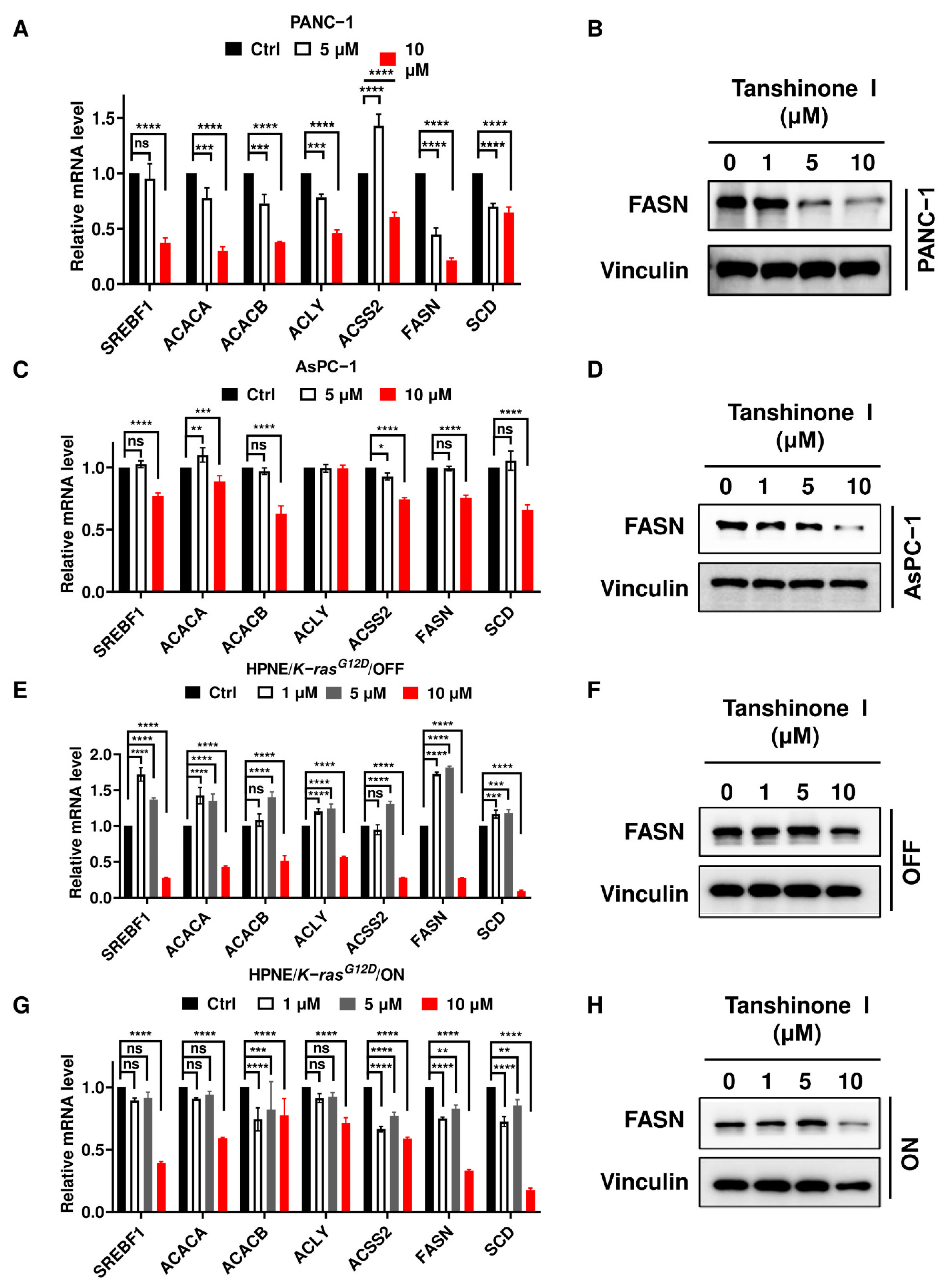
2.4. Inhibition of PLA2G2A Significantly Suppresses the Expression of FASN Involved in De Novo Fatty Acid Synthesis
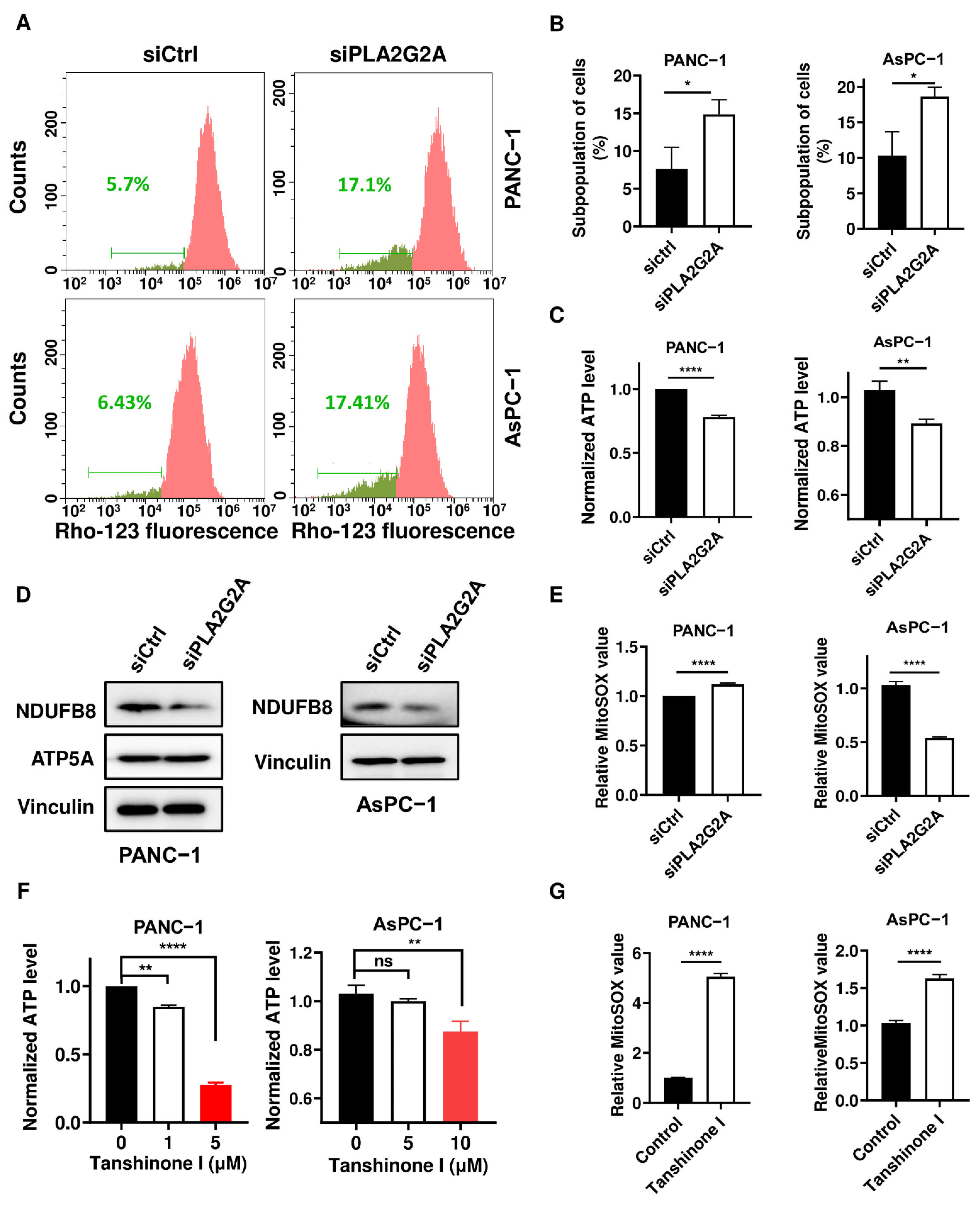
2.5. Inhibition of PLA2G2A Causes Mitochondria Dysfunction in PDAC Cells
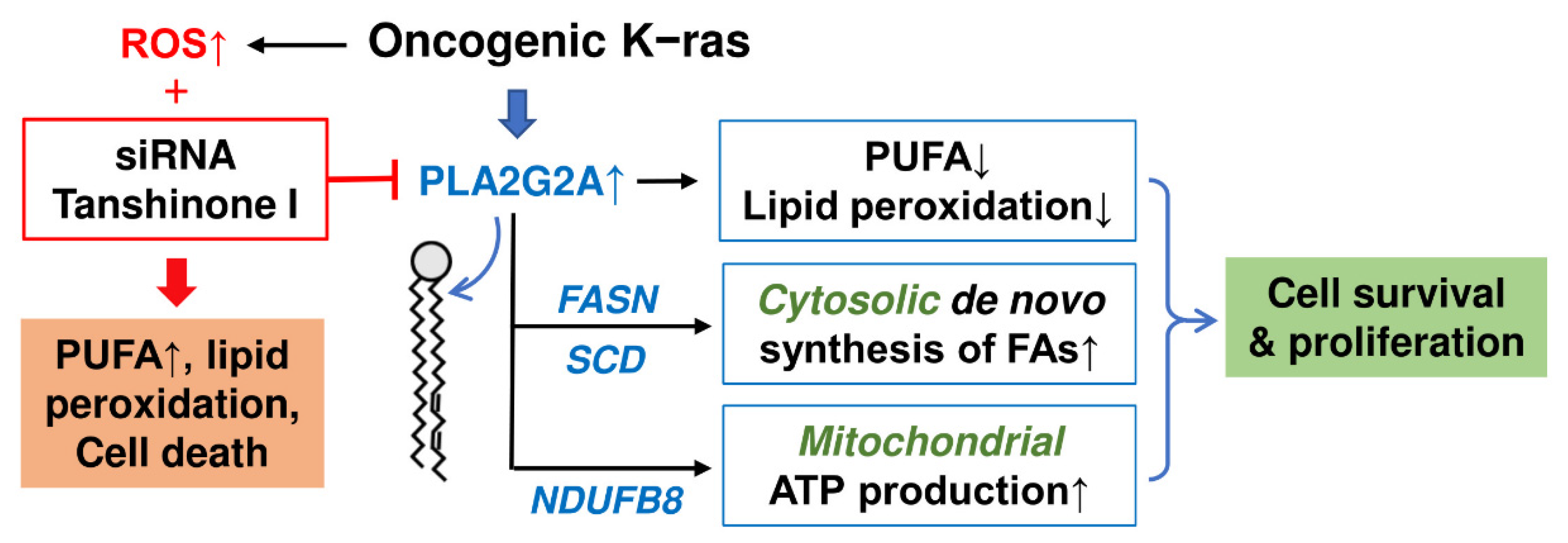
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Culture Reagents
4.3. Bioinformatics
4.4. Cell Transfection
4.5. MTT Assay
4.6. Colony Formation Assay
4.7. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.8. Western Blotting
4.9. Measurement of Cellular Mitochondrial ROS, Lipid Peroxidation, and Mitochondrial Transmembrane Potential
4.10. Cellular ATP Detection
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [Green Version]
- Christie, W.W.; Harwood, J.L. Oxidation of polyunsaturated fatty acids to produce lipid mediators. Essays Biochem. 2020, 64, 401–421. [Google Scholar] [CrossRef]
- Koundouros, N.; Karali, E.; Tripp, A.; Valle, A.; Inglese, P.; Perry, N.J.S.; Magee, D.J.; Anjomani Virmouni, S.; Elder, G.A.; Tyson, A.L.; et al. Metabolic Fingerprinting Links Oncogenic PIK3CA with Enhanced Arachidonic Acid-Derived Eicosanoids. Cell 2020, 181, 1596–1611.e1527. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Sanidad, K.Z.; Shih, P.A.; Zhao, X.; Zhang, G. Eicosanoid signaling in carcinogenesis of colorectal cancer. Cancer Metastasis Rev. 2018, 37, 257–267. [Google Scholar] [CrossRef]
- Panagiotopoulos, A.A.; Kalyvianaki, K.; Castanas, E.; Kampa, M. Eicosanoids in prostate cancer. Cancer Metastasis Rev. 2018, 37, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Dore, E.; Boilard, E. Roles of secreted phospholipase A(2) group IIA in inflammation and host defense. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Van Hensbergen, V.P.; Wu, Y.; van Sorge, N.M.; Touqui, L. Type IIA Secreted Phospholipase A2 in Host Defense against Bacterial Infections. Trends Immunol. 2020, 41, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Mauchley, D.; Meng, X.; Johnson, T.; Fullerton, D.A.; Weyant, M.J. Modulation of growth in human esophageal adenocarcinoma cells by group IIa secretory phospholipase A(2). J. Thorac. Cardiovasc. Surg. 2010, 139, 591–599, discussion 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, Y.C.; Dong, B.; Wei, W.Q.; He, Y.; Li, X.Q.; Cormier, R.T.; Wang, W.; Liu, F. Overexpression of phospholipase A2 Group IIA in esophageal squamous cell carcinoma and association with cyclooxygenase-2 expression. Asian Pac. J. Cancer Prev. 2014, 15, 9417–9421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.K.; Rostomily, R.; Wong, S.T.C. Prognostic Gene Discovery in Glioblastoma Patients using Deep Learning. Cancers 2019, 11, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormier, R.T.; Hong, K.H.; Halberg, R.B.; Hawkins, T.L.; Richardson, P.; Mulherkar, R.; Dove, W.F.; Lander, E.S. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat. Genet. 1997, 17, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Fijneman, R.J.; Bade, L.K.; Peham, J.R.; van de Wiel, M.A.; van Hinsbergh, V.W.; Meijer, G.A.; O’Sullivan, M.G.; Cormier, R.T. Pla2g2a attenuates colon tumorigenesis in azoxymethane-treated C57BL/6 mice; expression studies reveal Pla2g2a target genes and pathways. Cell Oncol. 2009, 31, 345–356. [Google Scholar] [CrossRef]
- Wang, X.; Huang, C.J.; Yu, G.Z.; Wang, J.J.; Wang, R.; Li, Y.M.; Wu, Q. Expression of group IIA phospholipase A2 is an independent predictor of favorable outcome for patients with gastric cancer. Hum. Pathol. 2013, 44, 2020–2027. [Google Scholar] [CrossRef]
- Xing, X.F.; Li, H.; Zhong, X.Y.; Zhang, L.H.; Wang, X.H.; Liu, Y.Q.; Jia, S.Q.; Shi, T.; Niu, Z.J.; Peng, Y.; et al. Phospholipase A2 group IIA expression correlates with prolonged survival in gastric cancer. Histopathology 2011, 59, 198–206. [Google Scholar] [CrossRef]
- Li, Z.M.; Xu, S.W.; Liu, P.Q. Salvia miltiorrhizaBurge (Danshen): A golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol. Sin. 2018, 39, 802–824. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Moon, T.C.; Chang, H.W.; Son, K.H.; Kang, S.S.; Kim, H.P. Effects of tanshinone I isolated from Salvia miltiorrhiza bunge on arachidonic acid metabolism and in vivo inflammatory responses. Phytother. Res. 2002, 16, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e775. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Meng, N.; Glorieux, C.; Zhang, Y.; Liang, L.; Zeng, P.; Lu, W.; Huang, P. Oncogenic K-ras Induces Mitochondrial OPA3 Expression to Promote Energy Metabolism in Pancreatic Cancer Cells. Cancers 2019, 12, 65. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhu, C.F.; Ma, M.Z.; Chen, G.; Song, M.; Zeng, Z.L.; Lu, W.H.; Yang, J.; Wen, S.; Chiao, P.J.; et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget 2015, 6, 21148–21158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Chakravarti, S. Differential expression of inflammatory and fibrogenic genes and their regulation by NF-kappaB inhibition in a mouse model of chronic colitis. J. Immunol. 2007, 179, 6988–7000. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, J. Tanshinone I induces cell apoptosis by reactive oxygen species-mediated endoplasmic reticulum stress and by suppressing p53/DRAM-mediated autophagy in human hepatocellular carcinoma. Artif. Cells Nanomed. Biotechnol. 2020, 48, 488–497. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, J.; Liang, Y.; Mu, W.; Hou, X.; Ma, X.; Cao, Q. Tanshinone l exhibits anticancer effects in human endometrial carcinoma HEC-1-A cells via mitochondrial mediated apoptosis, cell cycle arrest and inhibition of JAK/STAT signalling pathway. J. Buon 2018, 23, 1092–1096. [Google Scholar]
- Huang, X.; Jin, L.; Deng, H.; Wu, D.; Shen, Q.K.; Quan, Z.S.; Zhang, C.H.; Guo, H.Y. Research and Development of Natural Product Tanshinone I: Pharmacology, Total Synthesis, and Structure Modifications. Front. Pharmacol. 2022, 13, 920411. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, S.H.; Zhen, W.X.; Cheng, T.; Wang, D.; Lin, J.B.; Wu, Y.H.; Wang, Y.F.; Chen, Y.; Shu, L.P.; et al. Tanshinone I, a new EZH2 inhibitor restricts normal and malignant hematopoiesis through upregulation of MMP9 and ABCG2. Theranostics 2021, 11, 6891–6904. [Google Scholar] [CrossRef]
- Lee, C.Y.; Sher, H.F.; Chen, H.W.; Liu, C.C.; Chen, C.H.; Lin, C.S.; Yang, P.C.; Tsay, H.S.; Chen, J.J. Anticancer effects of tanshinone I in human non-small cell lung cancer. Mol. Cancer Ther. 2008, 7, 3527–3538. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gong, Y.; Li, L.; Abdolmaleky, H.M.; Zhou, J.R. Bioactive tanshinone I inhibits the growth of lung cancer in part via downregulation of Aurora A function. Mol. Carcinog. 2013, 52, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Xia, X.; You, X.; Wang, Z.; Han, Y.; Yang, J.; Noppe, G.; Meester, C.D.; Ling, J.; Robert, A.; et al. Cisplatin and gemcitabine exert opposite effects on immunotherapy with PD-1 antibody in K-ras-driven cancer. J. Adv. Res. 2022, 40, 109–124. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Xiang, R.; Glorieux, C.; Huang, P. PLA2G2A Phospholipase Promotes Fatty Acid Synthesis and Energy Metabolism in Pancreatic Cancer Cells with K-ras Mutation. Int. J. Mol. Sci. 2022, 23, 11721. https://doi.org/10.3390/ijms231911721
Zhang M, Xiang R, Glorieux C, Huang P. PLA2G2A Phospholipase Promotes Fatty Acid Synthesis and Energy Metabolism in Pancreatic Cancer Cells with K-ras Mutation. International Journal of Molecular Sciences. 2022; 23(19):11721. https://doi.org/10.3390/ijms231911721
Chicago/Turabian StyleZhang, Mingquan, Rong Xiang, Christophe Glorieux, and Peng Huang. 2022. "PLA2G2A Phospholipase Promotes Fatty Acid Synthesis and Energy Metabolism in Pancreatic Cancer Cells with K-ras Mutation" International Journal of Molecular Sciences 23, no. 19: 11721. https://doi.org/10.3390/ijms231911721
APA StyleZhang, M., Xiang, R., Glorieux, C., & Huang, P. (2022). PLA2G2A Phospholipase Promotes Fatty Acid Synthesis and Energy Metabolism in Pancreatic Cancer Cells with K-ras Mutation. International Journal of Molecular Sciences, 23(19), 11721. https://doi.org/10.3390/ijms231911721