
Nemaline Myopathy in Brazilian Patients: Molecular and Clinical Characterization
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Clinical Data
2.2. Molecular Data
2.2.1. Patients with Mutations in the Nebulin Gene
2.2.2. Patients with Mutations in the ACTA1 Gene
2.2.3. Patients with Mutations in Other Genes Associated with NM
2.3. Muscle Biopsy
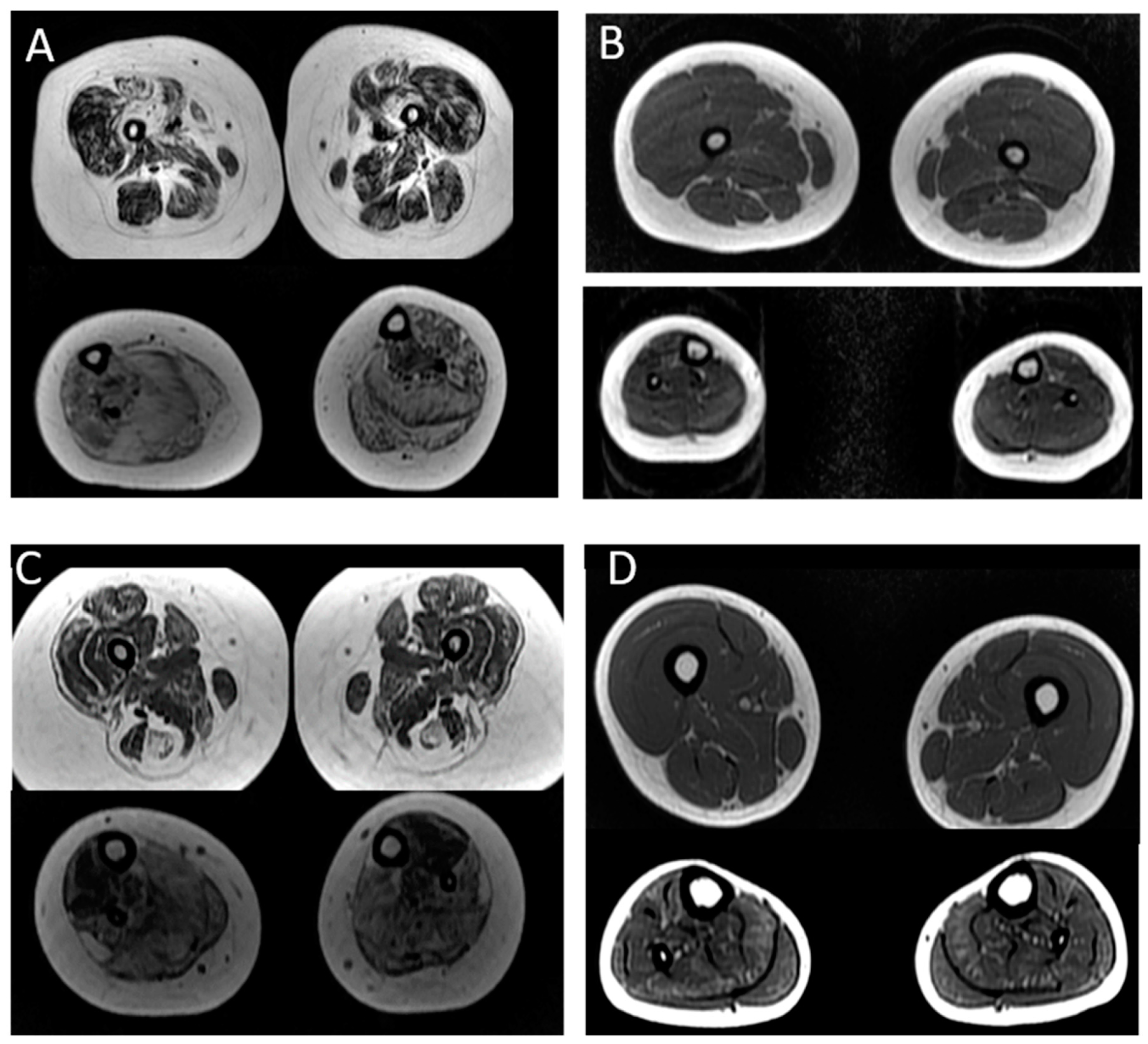
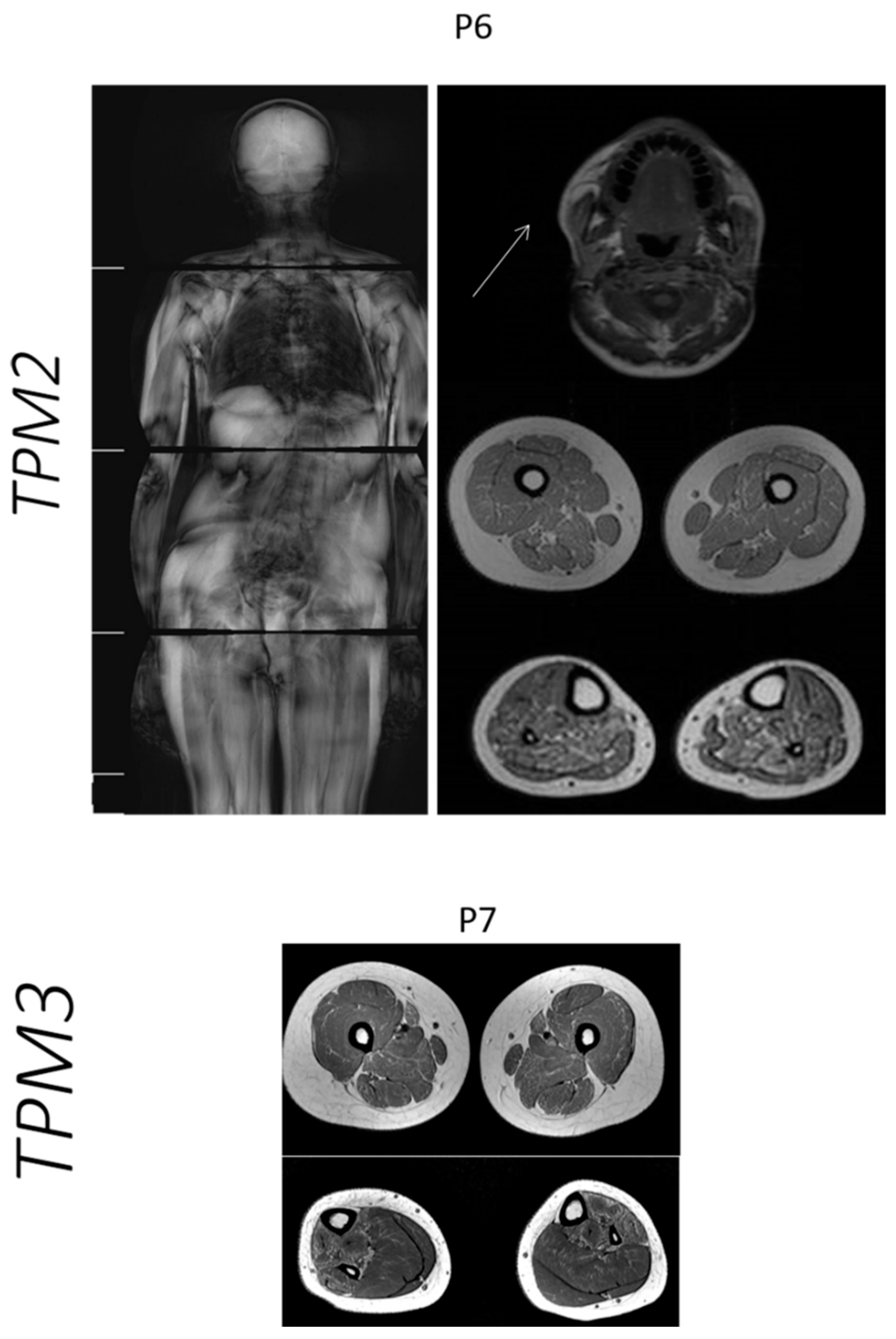
2.4. Muscle Imaging
3. Discussion
3.1. Nebulin Gene Mutations
3.2. ACTA1 Gene Mutations
3.3. Mutations in Other Genes Related to NM
3.4. Muscle Imaging in NM
4. Materials and Methods
4.1. Patients
4.2. Muscle Biopsy
4.3. Mutation Analysis
4.4. Muscle Imaging
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallgren-Pettersson, C.; Laing, N.G. Report of the 70th ENMC International Workshop: Nemaline Myopathy, 11–13 June 1999, Naarden, The Netherlands. Neuromuscul. Disord. 2000, 10, 299–306. [Google Scholar] [CrossRef]
- Romero, N.B.; Sandaradura, S.A.; Clarke, N.F. Recent Advances in Nemaline Myopathy. Curr. Opin. Neurol. 2013, 26, 519–526. [Google Scholar] [CrossRef]
- Malfatti, E.; Romero, N.B. Nemaline Myopathies: State of the Art. Rev. Neurol. 2016, 172, 614–619. [Google Scholar] [CrossRef]
- Dubowitz, V.; Sewry, C.A. Congenital Myopathies in Muscle Biopsy. A Practical Approach, 3rd ed.; Elsevier: Philadelphia, PA, USA, 2007. [Google Scholar]
- Laitila, J.; Wallgren-Pettersson, C. Recent Advances in Nemaline Myopathy. Neuromuscul. Disord. 2021, 31, 955–967. [Google Scholar] [CrossRef]
- Laing, N.G.; Majda, B.T.; Akkari, P.A.; Layton, M.G.; Mulley, J.C.; Phillips, H.; Haan, E.A.; White, S.J.; Beggs, A.H.; Kunkel, L.M.; et al. Assignment of a Gene (NEM1) for Autosomal Dominant Nemaline Myopathy to Chromosome I. Am. J. Hum. Genet. 1992, 50, 576–583. [Google Scholar]
- Laing, N.G.; Wilton, S.D.; Akkari, P.A.; Dorosz, S.; Boundy, K.; Kneebone, C.; Blumbergs, P.; White, S.; Watkins, H.; Love, D.R.; et al. A Mutation in the α Tropomyosin Gene TPM3 Associated with Autosomal Dominant Nemaline Myopathy. Nat. Genet. 1995, 9, 75–79. [Google Scholar] [CrossRef]
- Nowak, K.J.; Wattanasirichaigoon, D.; Goebel, H.H.; Wilce, M.; Pelin, K.; Donner, K.; Jacob, R.L.; Hübner, C.; Oexle, K.; Anderson, J.R.; et al. Mutations in the Skeletal Muscle α-Actin Gene in Patients with Actin Myopathy and Nemaline Myopathy. Nat. Genet. 1999, 23, 208–212. [Google Scholar] [CrossRef]
- Pelin, K.; Hilpelä, P.; Donner, K.; Sewry, C.; Akkari, P.A.; Wilton, S.D.; Wattanasirichaigoon, D.; Bang, M.-L.; Centner, T.; Hanefeld, F.; et al. Mutations in the Nebulin Gene Associated with Autosomal Recessive Nemaline Myopathy. Proc. Natl. Acad. Sci. USA 1999, 96, 2305–2310. [Google Scholar] [CrossRef] [Green Version]
- Donner, K.; Ollikainen, M.; Ridanpää, M.; Christen, H.-J.; Goebel, H.H.; de Visser, M.; Pelin, K.; Wallgren-Pettersson, C. Mutations in the β-Tropomyosin (TPM2) Gene—A Rare Cause of Nemaline Myopathy. Neuromuscul. Disord. 2002, 12, 151–158. [Google Scholar] [CrossRef]
- Johnston, J.J.; Kelley, R.I.; Crawford, T.O.; Morton, D.H.; Agarwala, R.; Koch, T.; Schäffer, A.A.; Francomano, C.A.; Biesecker, L.G. A Novel Nemaline Myopathy in the Amish Caused by a Mutation in Troponin T1. Am. J. Hum. Genet. 2000, 67, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, P.B.; Greenleaf, R.S.; Tomczak, K.K.; Lehtokari, V.-L.; Wallgren-Pettersson, C.; Wallefeld, W.; Laing, N.G.; Darras, B.T.; Maciver, S.K.; Dormitzer, P.R.; et al. Nemaline Myopathy with Minicores Caused by Mutation of the CFL2 Gene Encoding the Skeletal Muscle Actin–Binding Protein, Cofilin-2. Am. J. Hum. Genet. 2007, 80, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Sambuughin, N.; Yau, K.S.; Olivé, M.; Duff, R.M.; Bayarsaikhan, M.; Lu, S.; Gonzalez-Mera, L.; Sivadorai, P.; Nowak, K.J.; Ravenscroft, G.; et al. Dominant Mutations in KBTBD13, a Member of the BTB/Kelch Family, Cause Nemaline Myopathy with Cores. Am. J. Hum. Genet. 2010, 87, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravenscroft, G.; Miyatake, S.; Lehtokari, V.-L.; Todd, E.J.; Vornanen, P.; Yau, K.S.; Hayashi, Y.K.; Miyake, N.; Tsurusaki, Y.; Doi, H.; et al. Mutations in KLHL40 Are a Frequent Cause of Severe Autosomal-Recessive Nemaline Myopathy. Am. J. Hum. Genet. 2013, 93, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.A.; Ravenscroft, G.; Shaheen, R.; Todd, E.J.; Swanson, L.C.; Shiina, M.; Ogata, K.; Hsu, C.; Clarke, N.F.; Darras, B.T.; et al. Identification of KLHL41 Mutations Implicates BTB-Kelch-Mediated Ubiquitination as an Alternate Pathway to Myofibrillar Disruption in Nemaline Myopathy. Am. J. Hum. Genet. 2013, 93, 1108–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, M.; Sandaradura, S.A.; Dowling, J.J.; Kostyukova, A.S.; Moroz, N.; Quinlan, K.G.; Lehtokari, V.-L.; Ravenscroft, G.; Todd, E.J.; Ceyhan-Birsoy, O.; et al. Leiomodin-3 Dysfunction Results in Thin Filament Disorganization and Nemaline Myopathy. J. Clin. Investig. 2014, 124, 4693–4708. [Google Scholar] [CrossRef]
- Sandaradura, S.A.; North, K.N. LMOD3: The “Missing Link” in Nemaline Myopathy? Oncotarget 2015, 6, 26548–26549. [Google Scholar] [CrossRef]
- Malfatti, E.; Böhm, J.; Lacène, E.; Beuvin, M.; Romero, N.B.; Laporte, J. A Premature Stop Codon in MYO18B Is Associated with Severe Nemaline Myopathy with Cardiomyopathy. J. Neuromuscul. Dis. 2015, 2, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, S.; Mitsuhashi, S.; Hayashi, Y.K.; Purevjav, E.; Nishikawa, A.; Koshimizu, E.; Suzuki, M.; Yatabe, K.; Tanaka, Y.; Ogata, K.; et al. Biallelic Mutations in MYPN, Encoding Myopalladin, Are Associated with Childhood-Onset, Slowly Progressive Nemaline Myopathy. Am. J. Hum. Genet. 2017, 100, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Nilipour, Y.; Nafissi, S.; Tjust, A.E.; Ravenscroft, G.; Hossein Nejad Nedai, H.; Taylor, R.L.; Varasteh, V.; Pedrosa Domellöf, F.; Zangi, M.; Tonekaboni, S.H.; et al. Ryanodine Receptor Type 3 (RYR3) as a Novel Gene Associated with a Myopathy with Nemaline Bodies. Eur. J. Neurol. 2018, 25, 841–847. [Google Scholar] [CrossRef]
- Sandaradura, S.A.; Bournazos, A.; Mallawaarachchi, A.; Cummings, B.B.; Waddell, L.B.; Jones, K.J.; Troedson, C.; Sudarsanam, A.; Nash, B.M.; Peters, G.B.; et al. Nemaline Myopathy and Distal Arthrogryposis Associated with an Autosomal Recessive TNNT3 Splice Variant. Hum. Mutat. 2018, 39, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Nishikawa, A.; Iida, A.; Mori-Yoshimura, M.; Oya, Y.; Ishiyama, A.; Komaki, H.; Nakamura, S.; Fujikawa, S.; Kanda, T.; et al. ADSSL1 Myopathy Is the Most Common Nemaline Myopathy in Japan with Variable Clinical Features. Neurology 2020, 95, e1500–e1511. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, T.; Lornage, X.; Carlier, P.G.; Bassez, G.; Brochier, G.; Chanut, A.; Lacène, E.; Bui, M.-T.; Metay, C.; Oppermann, U.; et al. A Heterozygous Mutation in the Filamin C Gene Causes an Unusual Nemaline Myopathy With Ring Fibers. J. Neuropathol. Exp. Neurol. 2020, 79, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Madigan, N.N.; Polzin, M.J.; Cui, G.; Liewluck, T.; Alsharabati, M.H.; Klein, C.J.; Windebank, A.J.; Mer, G.; Milone, M. Filamentous Tangles with Nemaline Rods in MYH2 Myopathy: A Novel Phenotype. Acta Neuropathol. Commun. 2021, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Knipfer, M.; Huang, Q.-Q.; van Heerden, A.; Hsu, L.C.-L.; Gutierrez, G.; Quian, X.-L.; Stedman, H. Human Skeletal Muscle Nebulin Sequence Encodes a Blueprint for Thin Filament Architecture. J. Biol. Chem. 1996, 271, 4304–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtokari, V.-L.; Pelin, K.; Sandbacka, M.; Ranta, S.; Donner, K.; Muntoni, F.; Sewry, C.; Angelini, C.; Bushby, K.; van den Bergh, P.; et al. Identification of 45 Novel Mutations in the Nebulin Gene Associated with Autosomal Recessive Nemaline Myopathy. Hum. Mutat. 2006, 27, 946–956. [Google Scholar] [CrossRef]
- Lehtokari, V.-L.; Kiiski, K.; Sandaradura, S.A.; Laporte, J.; Repo, P.; Frey, J.A.; Donner, K.; Marttila, M.; Saunders, C.; Barth, P.G.; et al. Mutation Update: The Spectra of Nebulin Variants and Associated Myopathies. Hum. Mutat. 2014, 35, 1418–1426. [Google Scholar] [CrossRef] [Green Version]
- Kiiski, K.; Lehtokari, V.-L.; Löytynoja, A.; Ahlstén, L.; Laitila, J.; Wallgren-Pettersson, C.; Pelin, K. A Recurrent Copy Number Variation of the NEB Triplicate Region: Only Revealed by the Targeted Nemaline Myopathy CGH Array. Eur. J. Hum. Genet. 2016, 24, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Piga, D.; Magri, F.; Ronchi, D.; Corti, S.; Cassandrini, D.; Mercuri, E.; Tasca, G.; Bertini, E.; Fattori, F.; Toscano, A.; et al. New Mutations in NEB Gene Discovered by Targeted Next-Generation Sequencing in Nemaline Myopathy Italian Patients. J. Mol. Neurosci. 2016, 59, 351–359. [Google Scholar] [CrossRef]
- Jungbluth, H.; Sewry, C.A.; Counsell, S.; Allsop, J.; Chattopadhyay, A.; Mercuri, E.; North, K.; Laing, N.; Bydder, G.; Pelin, K.; et al. Magnetic Resonance Imaging of Muscle in Nemaline Myopathy. Neuromuscul. Disord. 2004, 14, 779–784. [Google Scholar] [CrossRef]
- Jarraya, M.; Quijano-Roy, S.; Monnier, N.; Béhin, A.; Avila-Smirnov, D.; Romero, N.B.; Allamand, V.; Richard, P.; Barois, A.; May, A.; et al. Whole-Body Muscle MRI in a Series of Patients with Congenital Myopathy Related to TPM2 Gene Mutations. Neuromuscul. Disord. 2012, 22, S137–S147. [Google Scholar] [CrossRef]
- Quijano-Roy, S.; Avila-Smirnow, D.; Carlier, R.-Y.; Bevilacqua, J.A.; Romero, N.B.; Fischer, D. Congenital Myopathies. In Neuromuscular Imaging; Springer: New York, NY, USA, 2013; pp. 147–176. [Google Scholar]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.H.; Sewry, C.; et al. Approach to the Diagnosis of Congenital Myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.C.; Nowak, K.J.; Durling, H.J.; Beggs, A.H.; Wallgren-Pettersson, C.; Romero, N.; Nonaka, I.; Laing, N.G. Muscle Disease Caused by Mutations in the Skeletal Muscle Alpha-Actin Gene (ACTA1). Neuromuscul. Disord. 2003, 13, 519–531. [Google Scholar] [CrossRef]
- Laing, N.G.; Dye, D.E.; Wallgren-Pettersson, C.; Richard, G.; Monnier, N.; Lillis, S.; Winder, T.L.; Lochmüller, H.; Graziano, C.; Mitrani-Rosenbaum, S.; et al. Mutations and Polymorphisms of the Skeletal Muscle Alpha-Actin Gene (ACTA1). Hum. Mutat. 2009, 30, 1267–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokbel, N.; Ilkovski, B.; Kreissl, M.; Memo, M.; Jeffries, C.M.; Marttila, M.; Lehtokari, V.-L.; Lemola, E.; Grönholm, M.; Yang, N.; et al. K7del Is a Common TPM2 Gene Mutation Associated with Nemaline Myopathy and Raised Myofibre Calcium Sensitivity. Brain 2013, 136, 494–507. [Google Scholar] [CrossRef] [Green Version]
- Clarke, N.F.; Kolski, H.; Dye, D.E.; Lim, E.; Smith, R.L.L.; Patel, R.; Fahey, M.C.; Bellance, R.; Romero, N.B.; Johnson, E.S.; et al. Mutations in TPM3 Are a Common Cause of Congenital Fiber Type Disproportion. Ann. Neurol. 2008, 63, 329–337. [Google Scholar] [CrossRef]
- Pelin, K.; Donner, K.; Holmberg, M.; Jungbluth, H.; Muntoni, F.; Wallgren-Pettersson, C. Nebulin Mutations in Autosomal Recessive Nemaline Myopathy: An Update. Neuromuscul. Disord. 2002, 12, 680–686. [Google Scholar] [CrossRef]
- Malfatti, E.; Lehtokari, V.-L.; Böhm, J.; de Winter, J.M.; Schäffer, U.; Estournet, B.; Quijano-Roy, S.; Monges, S.; Lubieniecki, F.; Bellance, R.; et al. Muscle Histopathology in Nebulin-Related Nemaline Myopathy: Ultrastrastructural Findings Correlated to Disease Severity and Genotype. Acta Neuropathol. Commun. 2014, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- Seferian, A.M.; Malfatti, E.; Bosson, C.; Pelletier, L.; Taytard, J.; Forin, V.; Gidaro, T.; Gargaun, E.; Carlier, P.; Fauré, J.; et al. Mild Clinical Presentation in KLHL40-Related Nemaline Myopathy (NEM 8). Neuromuscul. Disord. 2016, 26, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Piluso, G.; Dionisi, M.; del Vecchio Blanco, F.; Torella, A.; Aurino, S.; Savarese, M.; Giugliano, T.; Bertini, E.; Terracciano, A.; Vainzof, M.; et al. Motor Chip: A Comparative Genomic Hybridization Microarray for Copy-Number Mutations in 245 Neuromuscular Disorders. Clin. Chem. 2011, 57, 1584–1596. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Talim, B.; Moghadaszadeh, B.; Petit, N.; Brockington, M.; Counsell, S.; Guicheney, P.; Muntoni, F.; Merlini, L. Clinical and Imaging Findings in Six Cases of Congenital Muscular Dystrophy with Rigid Spine Syndrome Linked to Chromosome 1p (RSMD1). Neuromuscul. Disord. 2002, 12, 631–638. [Google Scholar] [CrossRef]
- Wattjes, M.P.; Kley, R.A.; Fischer, D. Neuromuscular Imaging in Inherited Muscle Diseases. Eur. Radiol. 2010, 20, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, H.; Treves, S.; Zorzato, F.; Sarkozy, A.; Ochala, J.; Sewry, C.; Phadke, R.; Gautel, M.; Muntoni, F. Congenital Myopathies: Disorders of Excitation–Contraction Coupling and Muscle Contraction. Nat. Rev. Neurol. 2018, 14, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Gurgel-Giannetti, J.; Reed, U.; Bang, M.-L.; Pelin, K.; Donner, K.; Marie, S.K.; Carvalho, M.; Fireman, M.A.T.; Zanoteli, E.; Oliveira, A.S.B.; et al. Nebulin Expression in Patients with Nemaline Myopathy. Neuromuscul. Disord. 2001, 11, 154–162. [Google Scholar] [CrossRef]
- Gurgel-Giannetti, J.; Bang, M.-L.; Reed, U.; Marie, S.; Zatz, M.; Labeit, S.; Vainzof, M. Lack of the C-terminal Domain of Nebulin in a Patient with Nemaline Myopathy. Muscle Nerve 2002, 25, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Gurgel-Giannetti, J.; Reed, U.C.; Marie, S.K.; Zanoteli, E.; Fireman, M.A.T.; Oliveira, A.S.B.; Werneck, L.C.; Beggs, A.H.; Zatz, M.; Vainzof, M. Rod Distribution and Muscle Fiber Type Modification in the Progression of Nemaline Myopathy. J. Child Neurol. 2003, 18, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Gonorazky, H.D.; Bönnemann, C.G.; Dowling, J.J. The Genetics of Congenital Myopathies. Handb. Clin. Neurol. 2018, 148, 549–564. [Google Scholar] [PubMed]
- Sasaki, M.; Yoneyama, H.; Nonaka, I. Respiratory Muscle Involvement in Nemaline Myopathy. Pediatr. Neurol. 1990, 6, 425–427. [Google Scholar] [CrossRef]
- Lindqvist, J.; Cheng, A.J.; Renaud, G.; Hardeman, E.C.; Ochala, J. Distinct Underlying Mechanisms of Limb and Respiratory Muscle Fiber Weaknesses in Nemaline Myopathy. J. Neuropathol. Exp. Neurol. 2013, 72, 472–481. [Google Scholar] [CrossRef] [Green Version]
- Joureau, B.; de Winter, J.M.; Stam, K.; Granzier, H.; Ottenheijm, C.A.C. Muscle Weakness in Respiratory and Peripheral Skeletal Muscles in a Mouse Model for Nebulin-Based Nemaline Myopathy. Neuromuscul. Disord. 2017, 27, 83–89. [Google Scholar] [CrossRef]
- Donner, K.; Sandbacka, M.; Lehtokari, V.-L.; Wallgren-Pettersson, C.; Pelin, K. Complete Genomic Structure of the Human Nebulin Gene and Identification of Alternatively Spliced Transcripts. Eur. J. Hum. Genet. 2004, 12, 744–751. [Google Scholar] [CrossRef]
- Pelin, K.; Wallgren-Pettersson, C. Nebulin-a Giant Chameleon. In The Sarcomere and Skeletal Muscle Disease; Springer: New York, NY, USA, 2008; Volume 642. [Google Scholar] [CrossRef]
- Yamamoto, D.L.; Vitiello, C.; Zhang, J.; Gokhin, D.S.; Castaldi, A.; Coulis, G.; Piaser, F.; Filomena, M.C.; Eggenhuizen, P.J.; Kunderfranco, P.; et al. The Nebulin SH3 Domain Is Dispensable for Normal Skeletal Muscle Structure but Is Required for Effective Active Load Bearing in Mouse. J. Cell Sci. 2013, 126, 5477–5489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtokari, V.-L.; Greenleaf, R.S.; DeChene, E.T.; Kellinsalmi, M.; Pelin, K.; Laing, N.G.; Beggs, A.H.; Wallgren-Pettersson, C. The Exon 55 Deletion in the Nebulin Gene–One Single Founder Mutation with World-Wide Occurrence. Neuromuscul. Disord. 2009, 19, 179–181. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Shin, J.; Kim, H.; Lee, C.; Kim, D. Characterization of Congenital Myopathies at a Korean Neuromuscular Center. Muscle Nerve 2018, 58, 235–244. [Google Scholar] [CrossRef]
- Lornage, X.; Quijano-Roy, S.; Amthor, H.; Carlier, R.-Y.; Monnier, N.; Deleuze, J.-F.; Romero, N.B.; Laporte, J.; Böhm, J. Asymmetric Muscle Weakness Due to ACTA1 Mosaic Mutations. Neurology 2020, 95, e3406–e3411. [Google Scholar] [CrossRef] [PubMed]
- Schreckenbach, T.; Schröder, J.M.; Voit, T.; Abicht, A.; Neuen-Jacob, E.; Roos, A.; Bulst, S.; Kuhl, C.; Schulz, J.B.; Weis, J.; et al. Novel TPM3 Mutation in a Family with Cap Myopathy and Review of the Literature. Neuromuscul. Disord. 2014, 24, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Naslavsky, M.S.; Yamamoto, G.L.; Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic Variants of an Elderly Cohort of Brazilians in the ABraOM Database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Case, Sex | Family | Clinical Phenotype | Molecular Analysis | |||
|---|---|---|---|---|---|---|
| Age at Last Evaluation and Clinical Findings | Gene | Mutations | Type/Segregation Parental | ACMG 6/2022 Classification | ||
| 1 Male | Family 1 | Typical 20 years: Not Ambulant since 11 years of age NIV at night, Scoliosis | ACTA1 | NM_001100.4:exon4:c.541G>C: p.(Asp181His) | Missense heterozygous Previously described [34] | Likely Pathogenic PM2 PP2 PP3 PS4_Supporting PP4 |
| 2 Male | Family 2 | Typical 13 years: Ambulant No VNI, No Scoliosis | ACTA1 | NM_001100.4:c.130-5T>A | Splice site Heterozygous Novel Not present in the parents | VUS PM2 PP2 PP3 PP4 |
| 3 Male | Family 3 | Severe 15 years: Not Ambulant (never) Permanent Invasive ventilation (tracheostomy) Gastrostomy, Scoliosis | ACTA1 | NM_001100.4:exon4:c.478G>A: p.(Gly160Ser) | Missense heterozygous Previously described [35] | Likely Pathogenic PM2 PP2 PP3 PS4_Supporting PP4 |
| 4 Male | Family 4 | Typical 3 years: Ambulant No VNI/ No scoliosis | ACTA1 | NM_001100.4:c.478G>A: p.(Arg198His) | Missense heterozygous Previously described [35] | Likely Pathogenic PM2 PP2 PP3 PS4_Supporting PP4 |
| 5 Female | Family 5 | Typical 14 years: Ambulant No VNI/ No scoliosis | ACTA1 | NM_001100.4:c.854T>G: p.(Met283Arg) | Missense heterozygous Previously described [35] | Likely Pathogenic PM2 PP2 PP3 PS4_Supporting PP4 |
| 6 Male | Family 6 | Typical 13 years: Ambulant No VNI/ Mild scoliosis | TPM2 | NM_003289.4:c.20_22 del: p.(Lys7del) | Frameshift heterozygous Previously described [36] | Pathogenic PM2 PS4 PP3 PM6 |
| 7 Male | Family 7 AD | Typical 19 years: Ambulant/Nocturnal VNI/Severe scoliosis | TPM3 | NM_152263.4:c.502C>T: p.(Arg168Cys) | Missense heterozygous Previously described [37] | Pathogenic PM2 PS4 PM5 PM6 |
| 8 Male (father) | Typical 62 years: Ambulant with support Permanent VNI (>16 h/day), Scoliosis | NM_152263.4:c.502C>T: p.(Arg168Cys) | ||||
| 9 Female | Family 8 | Mild 27 years: Ambulant No VNI | NEB | NM_001164507.1:c.8501delA: p.(Lys2834ArgfsTer28) | Frameshift heterozygous Novel | Pathogenic PVS1 PM2 PP4 |
| NM_001164507.1:c.1674+2T>C | Splice site heterozygous Previously described [27] | Pathogenic PVS1 PM2 PS4_Supporting PP4 | ||||
| 10 Male | Family 9 | Typical Dead at 25 years old: Not ambulant Permanent invasive ventilation (tracheostomy) Severe scoliosis | NEB | NM_001164507.1:exon170:c.24189_24192dup p.(Glu8065SerfsTer5) | Frameshift heterozygous Previously described [38] | Pathogenic PVS1 PM2 PS4_Supporting PP4 PP4 |
| NM_001164507.1:exon134:c.20466+2T>A | Splice site heterozygous Novel | Pathogenic PVS1 PM2 PM3_Supporting PP4 | ||||
| 11 Female | Family 10 consanguineous | Typical 4 years: Ambulant, No VNI | NEB | NM_001164507.1:exon172:c.24304_24305dup: p.(Leu8102PhefsTer44) | Frameshift homozygous Novel | Pathogenic PVS1 PM2 PP4 |
| 12 Male (brother) | Typical 6 years: Ambulant, No VNI | NM_001164507.1:exon172:c.24304_24305dup: p.(Leu8102PhefsTer44) | Frameshift homozygous Novel | Pathogenic PVS1 PM2 PP4 | ||
| 13 Male | Family 11 | Typical 14 years: Ambulant/ No VNI | NEB | NM_001164507.1:exon 63 c.8889+1G>A p.(?) | Splice site heterozygous Previously described [27] | Pathogenic PVS1 PM2 PS4_Moderate PP4 |
| NM_001164507.1:c.6869_6870insTGC: p.(Ala2290dup) | Non-frameshift duplication heterozygous Novel | VUS PM2 PM3_Supporting PP4 | ||||
| 14 Female | Family 12 | Typical 19 years: Ambulant Nocturnal NIV Scoliosis | NEB | NM_001164507.1:exon151:c.22170G>A: (p.Tyr7390Ter | Stop-gain Heterozygous Previously described [38]/maternal | Pathogenic PVS1 PM2 PS4_Supporting PM3_Supporting PP4 |
| NM_001164507.1:exon174: c.24579G>C; p.(Ser8193Ser) | Splicing heterozygous Previously described [39]/paternal | Likely Pathogenic PM2 PS4_Supporting PP3 PM3_Supporting PP4 | ||||
| 15 Female | Family 13 | Typical 23 years: Ambulant Permanent NIV (> 16 h a day) Scoliosis | NEB | NM_001164507.1:exon176:c.24735_24736del: p.(Arg8245fsTer2) | Frameshift deletion heterozygous Previously described [27,39] | Pathogenic PVS1 PM2 PS4_Supporting PP4 |
| NEB_NM_001164507.1:IVS122:c.19102-8_19102-4del | Splice site heterozygous Novel | VUS PM2 PP4 | ||||
| 16 Female | Family 14 | Typical 25 years: Lost the ambulation at 17 years of age Permanent NIV (>16 h a day) Severe Scoliosis | NEB | NM_001164507.1:exon174: c.24579G>C; p.(Ser8193Ser) | Splice site heterozygous Previously described [39]/paternal | Likely Pathogenic PM2 PS4_Supporting PP3 PM3_Supporting PP4 |
| NM_001164507.1:exon48:c.6078delA p.(Lys2026fs) | Frameshift heterozygous Previously described [26]/Maternal | Pathogenic PVS1, PM2 PS4_Supporting PP4 PM3_Supporting | ||||
| 17 Female | Family 15 | Typical 20 years: Ambulant Nocturnal NIV Scoliosis | NEB | NM_001164507.1:exon151:c.22170G>A: p.(Tyr7390Ter) | Stop-gain heterozygous Previously described [38]—nonsense mutation/paternal | Pathogenic PVS1 PM2 PS4_Supporting PP4 |
| NM_001164507.1:exon174: c.24579G>C; p.(Ser8193Ser) | Splice site heterozygous Previously described [39]/maternal | Likely Pathogenic PM2 PS4_Supporting PP3 PM3_Supporting PP4 | ||||
| 18 Female | Family 16 | Typical 4 years: Ambulant No NIV | NEB | NM_001164507.1: exon173: c.23601_23602del: p.(Lys7867AsnfsTer3) | Frameshift heterozygous NM consortium Previously described [38] | Pathogenic PVS1 PM2 PP4 |
| 19 Female (sister) | Typical 2 years: Ambulant No NIV | NM_001164507.1Int28:c.2835+5G>C | Splice site heterozygous NM consortium | VUS PM2 PP4 | ||
| 20 Female | Family 17 | Typical Dead at 15 years: Ambulant No NIV Acute respiratory failure | NEB | NM_001164507.1:c.23878_23881dup p.(Thr7961AsnfsTer16) | Frameshift heterozygous Novel | Pathogenic PVS1 PM2 PP4 |
| NM_001164507.1:c.25405-1G>C | Splice site heterozygous Novel | Pathogenic PVS1 P M2 PP4 | ||||
| 21 Female | Family 18 | Typical 12 years: Not ambulant Permanent NIV (had tracheostomy) Gastrostomy Scoliosis | NEB | NM_001164507.1:c.1623delT: p.(Asp541IlefsTer15) | Frameshift heterozygous Previously described [26]/maternal | Pathogenic PVS1 PM2 PS4_Supporting PP4 |
| Motor Chip (V. Nigro): duplicação 82 a 105 | CNV heterozygous Novel | |||||
| 22 Male | Family 19 | Typical 13 years: Ambulant No NIV | NEB | NM_001164507.1:exon34:c.3648del p.(Lys1218ArgfsTer6) | Frameshift heterozygous Novel | Pathogenic PVS1 PM2 PP4 |
| del exon 29 | CNV Novel Not confirmed by Motor Chip | Pathogenic PVS1 PM2 PP4 | ||||
| 23 Male | Family 20 | Mild 27 years: Ambulant No VNI | NEB | NM_001164507:exon43:c.5343+5G>A | Splicing homozygous Previously described [26] | Likely pathogenic PM2 (rara) PS4_sup PM4, PM3_sup, PP4 |
| 24 Female (sister) | Mild 25 years: Ambulant No VNI | NM_001164507:exon43:c.5343+5G>A | Splicing homozygous Previously described [26] | Likely pathogenic PM2 (rara) PS4_sup PM4, PM3_sup, PP4 | ||
| 25 Male | Family 21 | Typical 8 years: Not ambulant (never walked) Progressive weakness (loss of head control and the capacity to sit) Permanent NIV (>16 h a day) Gastrostomy Severe scoliosis | NEB | NM_001164507.1:c.21076C>T p.Arg7026Ter | Stop-gain heterozygous Previously reported [27] | Pathogenic PVS1 PM2 PS4_Moderate PP4 |
| NM_001164507.1:c.24192_24193insTCAA p.Glu8065Serfs5 | Frameshift heterozygous Previously reported [27] | Pathogenic PVS1 PM2 PS4 PM3 PP4 | ||||
| 26 Male | Family 22 | Typical Dead at 5 years old: Ambulant No NIV (Acute respiratory failure) | NEB | NM_001164507.1:exon129:c.19944G>A: p.(Ser6648Ser) | Splice site heterozygous Previously reported [27] | Likely Pathogenic PM2 PS4 PP3 PP4 |
| NM_001164507.1:exon105:c.16423A>T: p.(Lys5475Ter) | Stop-gain heterozygous Novel | Pathogenic PVS1 PM2 PP4 | ||||
| 27 Female | Family 23 | Typical 18 years: Ambulant No NIV No scoliosis | NEB | NM_001164507:exon43:c.5343+5G>A | Splice site heterozygous Previously reported [26] | Likely Pathogenic PM2 PS4_sup PM4 PM3_sup PP4 |
| 28 Male (brother) | Typical 13 years: Ambulant No NIV No scoliosis | NEB:NM_001164507:exon29:c.2943G>A p.Glu981Glu | Splice site heterozygous Previously reported by Invitae on Clinvar rs398124170 | Likely pathogenic PM2 PS4_sup PP3 PM3 PP4 | ||
| 29 Female | Family 24 | Severe 1,5 years: sits with support Nocturnal VNI Scoliosis | KLHL40 | NM_152393.4:c.1405G>A: p.(Gly469Ser) | Missense heterozygous Previously reported [14]/paternal | VUS PM2 PP3 PP4 |
| NM_152393.4:c.1498C>T: p.(Arg500Cys) | Missense heterozygous Previously reported [40]/maternal | VUS PM2 PS4_Supporting PP3 PP4 | ||||
| 30 Female | Family 25 | Severe 5 years: Not ambulant Distal Arthrogryposis Nocturnal VNI Scoliosis | KLHL40 | NM_152393.4:c.1405G>A: p.(Gly469Ser) | Missense heterozygous Previously reported [14] | VUS PM2 PP3 PP4 |
| NM_152393.4:c.1498C>T: p.(Arg500Cys) | Missense heterozygous Previously reported [40] | VUS PM2 PS4_Supporting PP3 PP4 | ||||
| Case | Age at Muscle MRI | Gene | Muscle MRI or CT (Mercuri Grade *) | |
|---|---|---|---|---|
| Thigh: | Legs: | |||
| P2 | 4 years | ACTA1 | No involvement | Tibialis anterior muscle: grade IIa Soleus muscle: no involvement |
| P3 | 8 years | ACTA1 | Diffuse fat infiltration of thigh muscles: grade III Biceps femoris and vastus lateralis muscles: grade IIb | Diffuse fat infiltration: grade III Tibialis posterior muscle: grade IIb |
| P6 | 12 years | TPM2 | Vastus lateralis and rectus femoris muscles: grade I OBS: Axial T1 image of the head: Involvement of temporal, masseter and pterygoid lateral muscles: grade II | Soleus muscle: grade IIb Gastrocnemius muscle: grade IIa |
| P7 | 13 years | TPM3 | Sartorius muscle: grade IIb Adductor magnus muscle: grade IIa Hamstring muscles: grade I | Soleus muscles: grade I Gastrocnemius muscle: grade I Tibialis anterior muscle: grade IIb Tibialis posterior muscle: grade IIb |
| P14 | 17 years | NEB | Diffuse fat infiltration of thigh muscles: grade I | Tibialis anterior muscle: grade III Soleus muscle: grade IIa |
| P15 | 15 years | NEB | Diffuse fat infiltration of thigh muscles: grade I | Tibialis anterior muscle: grade IIa Soleus muscle: grade IIb Fibular muscle: grade IIa |
| P16 | 17 years | NEB | Adductor longus muscle: grade III Adductor magnus muscle: grade III Rectus femoris muscle: grade IIa Vastus lateralis muscle: grade IIa | Tibialis anterior muscle: grade IV Soleus muscle: grade IV Fibular muscle: grade IV |
| P17 | 14 years | NEB | Diffuse fat infiltration of thigh muscles: grade I | Tibialis anterior muscle: grade II Soleus muscle: grade II Fibular. muscle: grade II |
| P21 | 7 years | NEB | Adductor longus muscle: grade I Adductor magnus muscle: grade IIb Rectus femoris muscle: grade IIb Vastus lateralis muscle: grade IIb | Tibialis anterior muscle: grade IIa Tibialis posterior muscle: grade IIa Soleus muscle: grade III Gastrocnemius muscle: grade III Fibular muscle: grade III |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurgel-Giannetti, J.; Souza, L.S.; Yamamoto, G.L.; Belisario, M.; Lazar, M.; Campos, W.; Pavanello, R.d.C.M.; Zatz, M.; Reed, U.; Zanoteli, E.; et al. Nemaline Myopathy in Brazilian Patients: Molecular and Clinical Characterization. Int. J. Mol. Sci. 2022, 23, 11995. https://doi.org/10.3390/ijms231911995
Gurgel-Giannetti J, Souza LS, Yamamoto GL, Belisario M, Lazar M, Campos W, Pavanello RdCM, Zatz M, Reed U, Zanoteli E, et al. Nemaline Myopathy in Brazilian Patients: Molecular and Clinical Characterization. International Journal of Molecular Sciences. 2022; 23(19):11995. https://doi.org/10.3390/ijms231911995
Chicago/Turabian StyleGurgel-Giannetti, Juliana, Lucas Santos Souza, Guilherme L. Yamamoto, Marina Belisario, Monize Lazar, Wilson Campos, Rita de Cassia M. Pavanello, Mayana Zatz, Umbertina Reed, Edmar Zanoteli, and et al. 2022. "Nemaline Myopathy in Brazilian Patients: Molecular and Clinical Characterization" International Journal of Molecular Sciences 23, no. 19: 11995. https://doi.org/10.3390/ijms231911995
APA StyleGurgel-Giannetti, J., Souza, L. S., Yamamoto, G. L., Belisario, M., Lazar, M., Campos, W., Pavanello, R. d. C. M., Zatz, M., Reed, U., Zanoteli, E., Oliveira, A. B., Lehtokari, V. -L., Casella, E. B., Machado-Costa, M. C., Wallgren-Pettersson, C., Laing, N. G., Nigro, V., & Vainzof, M. (2022). Nemaline Myopathy in Brazilian Patients: Molecular and Clinical Characterization. International Journal of Molecular Sciences, 23(19), 11995. https://doi.org/10.3390/ijms231911995