
Reduced DNAJC3 Expression Affects Protein Translocation across the ER Membrane and Attenuates the Down-Modulating Effect of the Translocation Inhibitor Cyclotriazadisulfonamide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
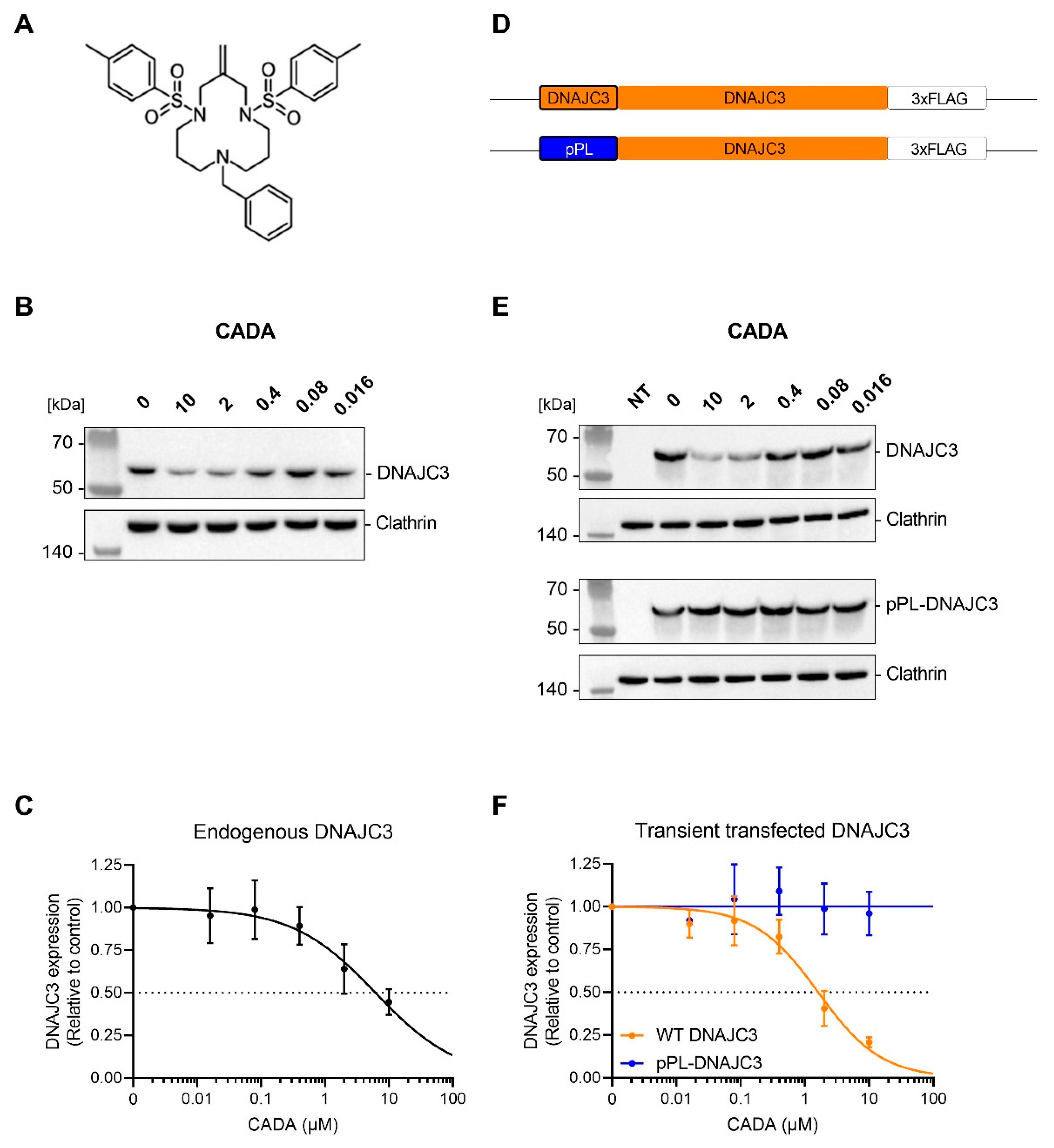
2.1. CADA Reduces Cellular Expression of DNAJC3
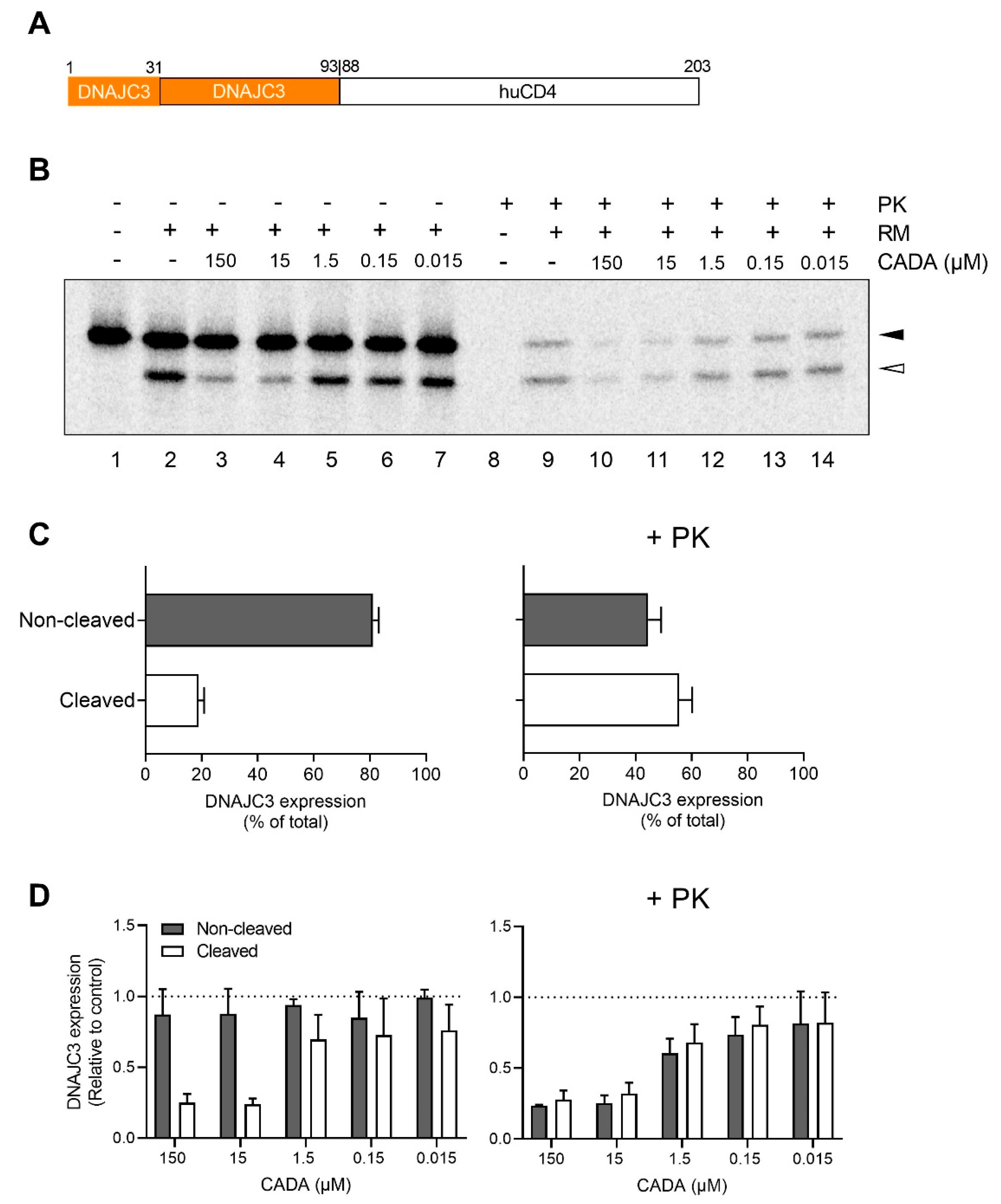
2.2. CADA Inhibits the Signal Peptide Dependent Co-Translational Translocation of the ER Lumenal DNAJC3 Protein
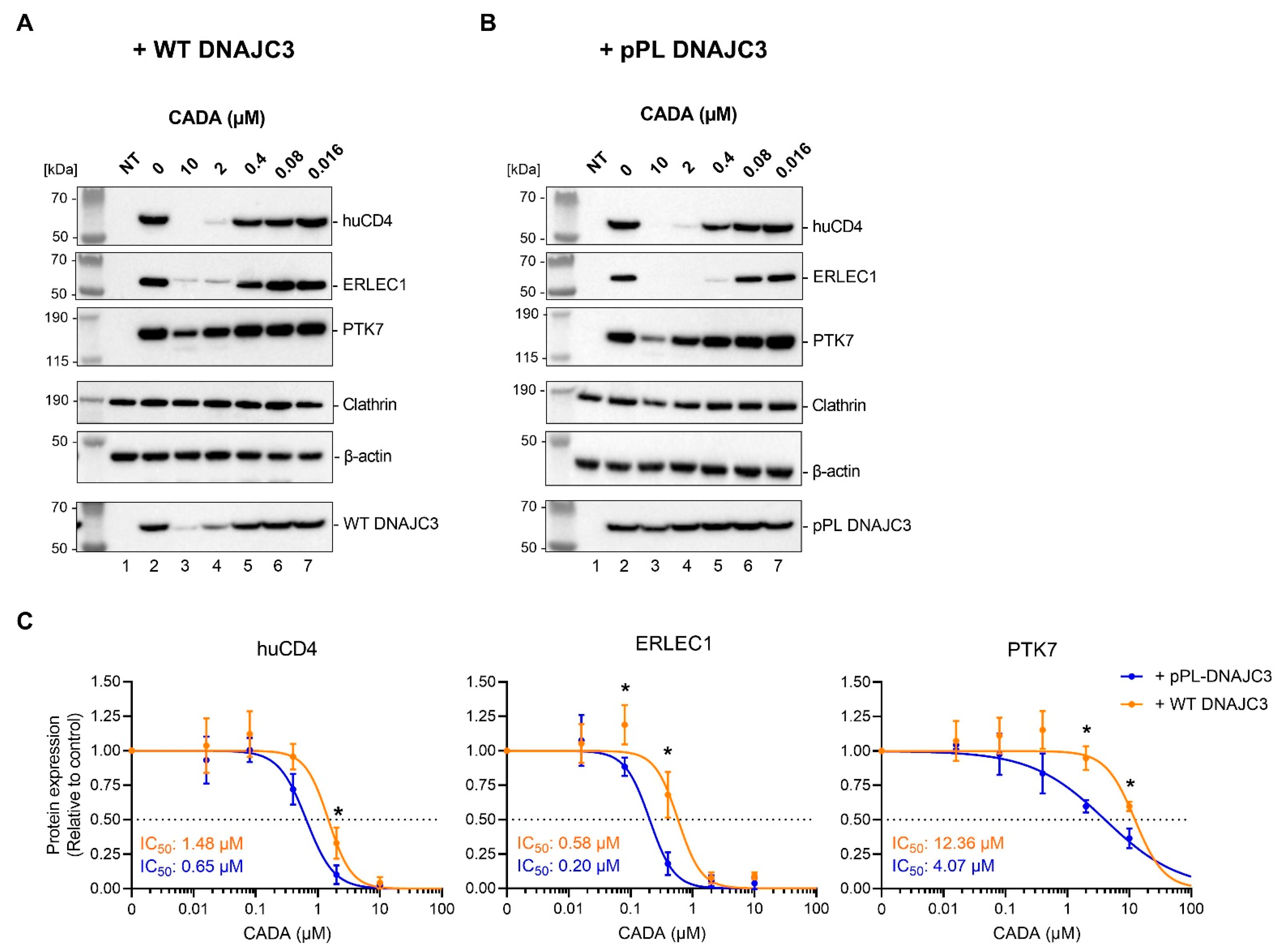
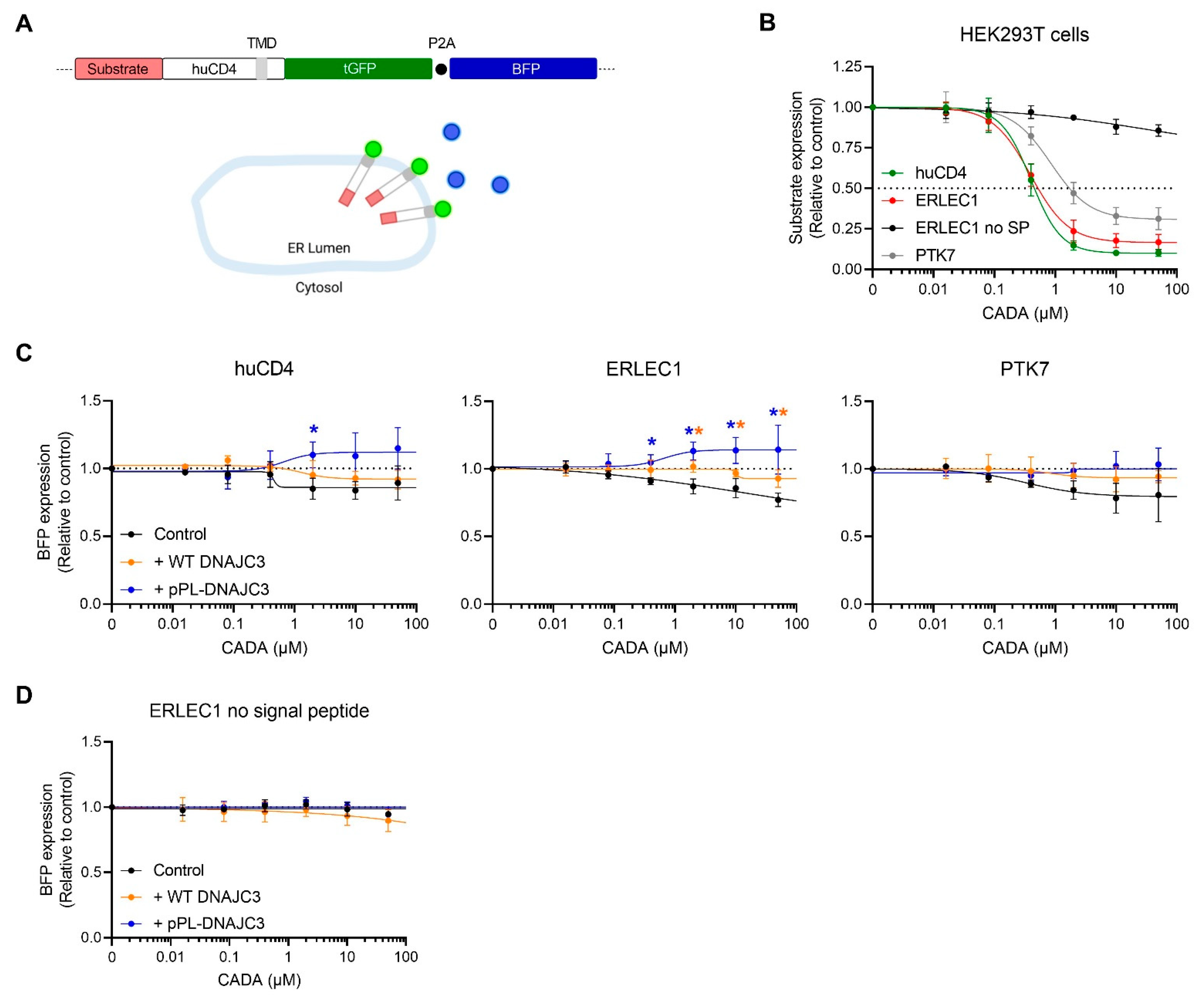
2.3. Presence of Cellular DNAJC3 Enhances the Sensitivity of Substrates to CADA
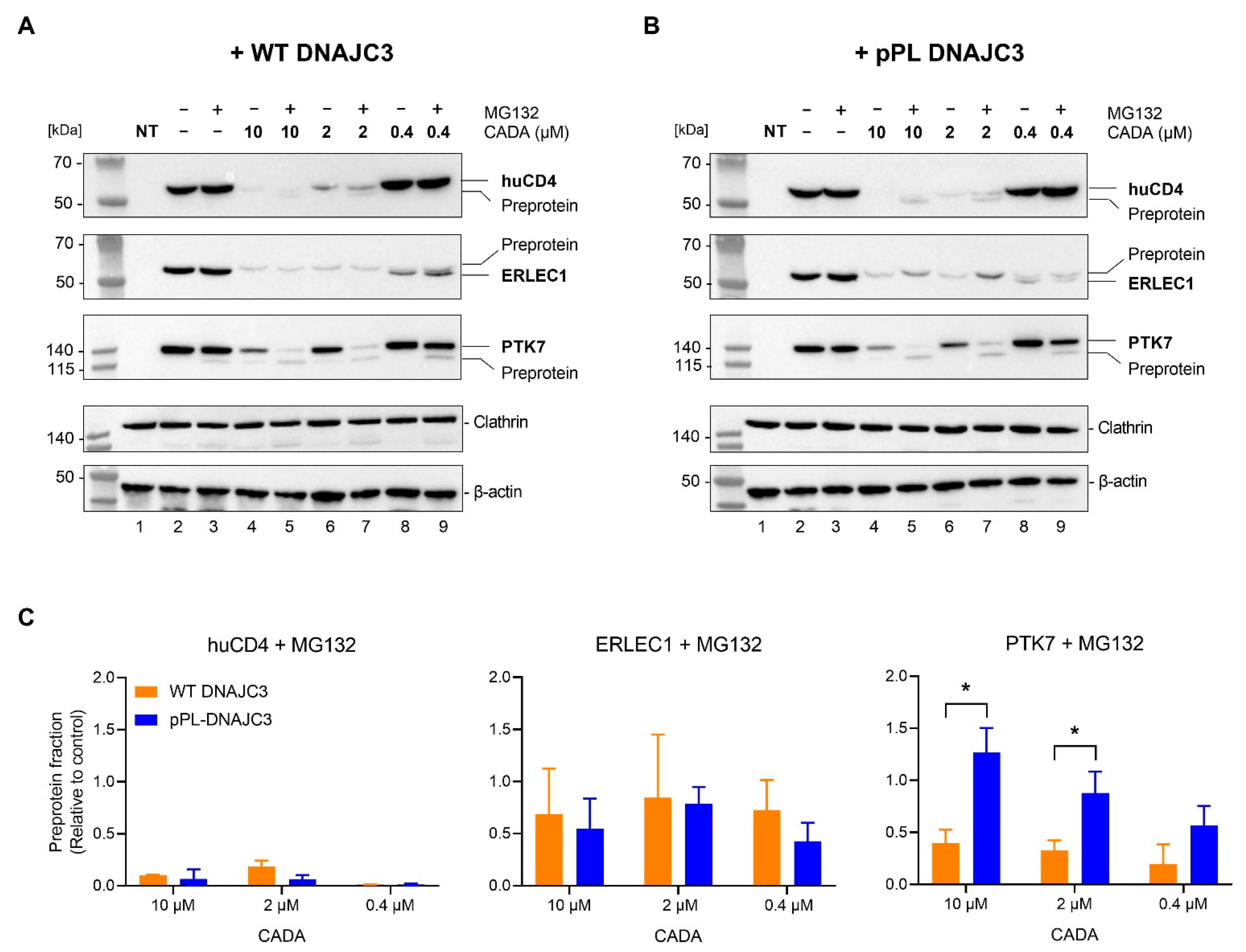
2.4. DNAJC3 Enhances Proteasomal Degradation of CADA-Stalled PTK7 Preprotein
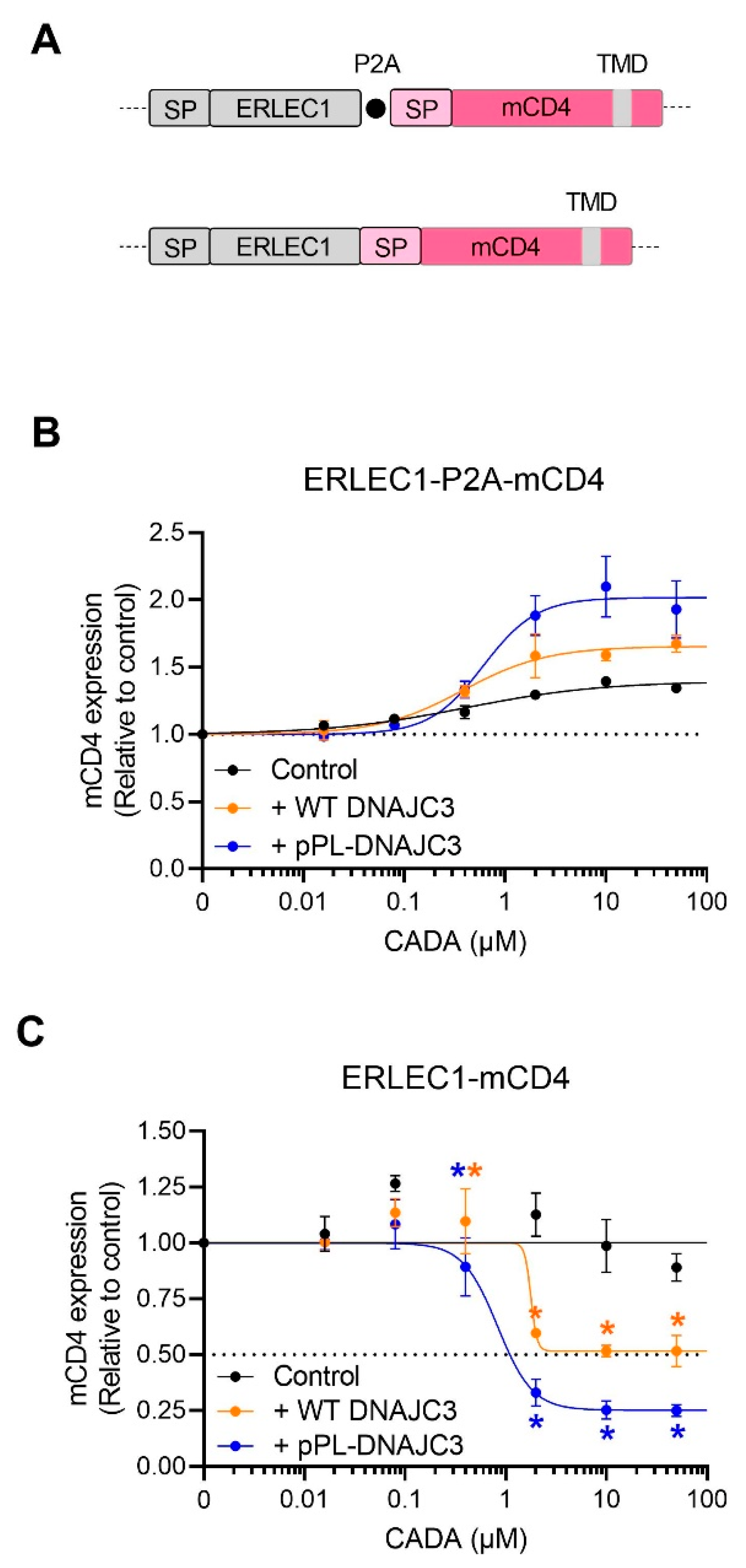
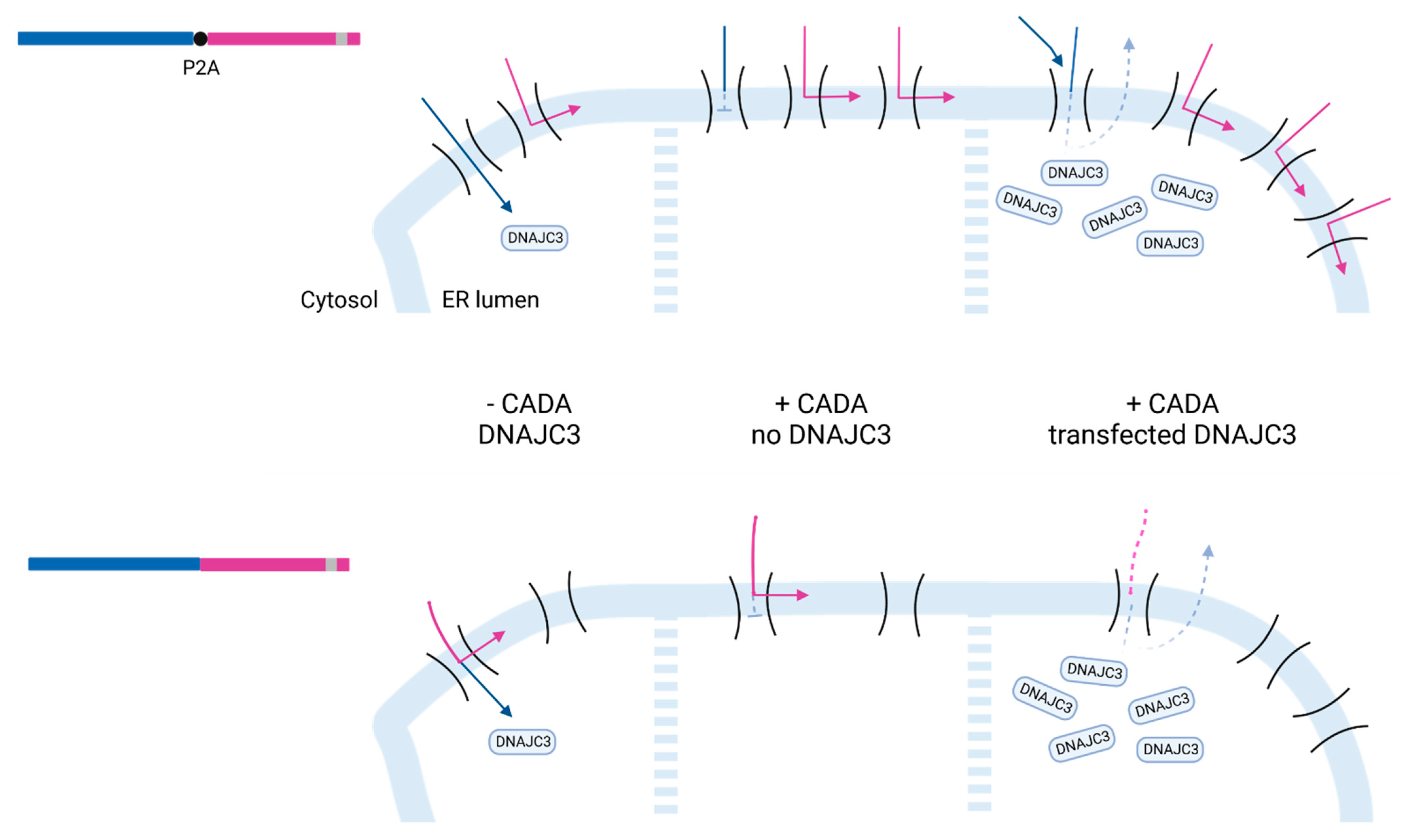
2.5. DNAJC3 Differentially Affects the Expression of Cytosolic or ER Translocated Proteins in the Presence of CADA-Stalled Substrates
3. Discussion
4. Materials and Methods
4.1. Compounds and Antibodies
4.2. Plasmids and Mutagenesis
4.3. Transient Transfection
4.4. Immunoblotting
4.5. Antibody Staining and Flow Cytometry
4.6. Cell-Free In Vitro Translation and Translocation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wickner, W.; Schekman, R. Protein translocation across biological membranes. Science 2005, 310, 1452–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, R.; Eyrisch, S.; Ahmad, M.; Helms, V. Protein translocation across the ER membrane. Biochim. Biophys. Acta 2011, 1808, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef]
- Dudek, J.; Pfeffer, S.; Lee, P.-H.; Jung, M.; Cavalié, A.; Helms, V.; Foerster, F.; Zimmermann, R. Protein Transport into the Human Endoplasmic Reticulum. J. Mol. Biol. 2015, 427, 1159–1175. [Google Scholar] [CrossRef] [PubMed]
- Aviram, N.; Schuldiner, M. Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J. Cell Sci. 2017, 130, 4079–4085. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Gilmore, R.; Blobel, G. Protein translocation across the endoplasmic reticulum. Cell 1984, 38, 5–8. [Google Scholar] [CrossRef]
- Pfeffer, S.; Burbaum, L.; Unverdorben, P.; Pech, M.; Chen, Y.; Zimmermann, R.; Beckmann, R.; Foerster, F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 8403. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, R.M.; Fernández, I.S.; Scheres, S.; Hegde, R.S. Structure of the Mammalian Ribosome-Sec61 Complex to 3.4 A Resolution. Cell 2014, 157, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorhees, R.M.; Hegde, R.S. Toward a structural understanding of co-translational protein translocation. Curr. Opin. Cell Biol. 2016, 41, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Brandt, F.; Hrabe, T.; Lang, S.; Eibauer, M.; Zimmermann, R.; Förster, F. Structure and 3D Arrangement of Endoplasmic Reticulum Membrane-Associated Ribosomes. Structure 2012, 20, 1508–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassdenteufel, S.; Nguyen, D.; Helms, V.; Lang, S.; Zimmermann, R. ER import of small human presecretory proteins: Components and mechanisms. FEBS Lett. 2019, 593, 2506–2524. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Powis, K.; High, S. Post-translational translocation into the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2403–2409. [Google Scholar] [CrossRef]
- Lakkaraju, A.K.; Thankappan, R.; Mary, C.; Garrison, J.L.; Taunton, J.; Strub, K. Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Mol. Biol. Cell 2012, 23, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Denks, K.; Vogt, A.; Sachelaru, I.; Petriman, N.A.; Kudva, R.; Koch, H.-G. The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol. Membr. Biol. 2014, 31, 58–84. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Haßdenteufel, S.; Theis, M.; Paton, A.W.; Paton, J.C.; Zimmermann, R.; High, S. The Signal Sequence Influences Post-Translational ER Translocation at Distinct Stages. PLoS ONE 2013, 8, e75394. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, R.M.; Hegde, R.S. Structure of the Sec61 channel opened by a signal sequence. Science 2016, 351, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Gemmer, M.; Förster, F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020, 133, jcs231340. [Google Scholar] [CrossRef]
- Hegde, R.S.; Kang, S.W. The concept of translocational regulation. J. Cell Biol. 2008, 182, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Foerster, F. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunger, K.; Pfeffer, S.; Shrimal, S.; Gilmore, R.; Berninghausen, O.; Mandon, E.C.; Becker, T.; Förster, F.; Beckmann, R. Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 2018, 350, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [Green Version]
- Mori, K. Tripartite Management of Unfolded Proteins in the Endoplasmic Reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ye, Y. Clearing Traffic Jams During Protein Translocation Across Membranes. Front. Cell Dev. Biol. 2020, 8, 610689. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999, 339 Pt 1, 135–141. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Brostrom, C.O.; Brostrom, M.A. Regulation of Translational Initiation during Cellular Responses to Stress. Prog. Nucleic Acid Res. Mol. Biol. 1997, 58, 79–125. [Google Scholar]
- Van Huizen, R.; Martindale, J.L.; Gorospe, M.; Holbrook, N.J. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J. Biol. Chem. 2003, 278, 15558–15564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, D.T.; Kang, S.-W.; Goodman, A.G.; Garrison, J.L.; Taunton, J.; Katze, M.G.; Kaufman, R.J.; Hegde, R.S. The Role of p58IPK in Protecting the Stressed Endoplasmic Reticulum. Mol. Biol. Cell 2007, 18, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2 kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [Green Version]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Sood, R.; Porter, A.C.; Ma, K.; Quilliam, L.A.; Wek, R.C. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem. J. 2000, 346 Pt 2, 281–293. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef]
- Jennings, M.J.; Hathazi, D.; Nguyen, C.D.L.; Munro, B.; Münchberg, U.; Ahrends, R.; Schenck, A.; Eidhof, I.; Freier, E.; Synofzik, M.; et al. Intracellular Lipid Accumulation and Mitochondrial Dysfunction Accompanies Endoplasmic Reticulum Stress Caused by Loss of the Co-chaperone DNAJC3. Front. Cell Dev. Biol. 2021, 9, 710247. [Google Scholar] [CrossRef]
- Roussel, B.D.; Kruppa, A.; Miranda, E.; Crowther, D.; Lomas, D.; Marciniak, S. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2018, 12, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Anttonen, A.K.; Mahjneh, I.; Hämäläinen, R.H.; Lagier-Tourenne, C.; Kopra, O.; Waris, L.; Anttonen, M.; Joensuu, T.; Kalimo, H.; Paetau, A.; et al. The gene disrupted in Marinesco-Sjögren syndrome encodes SIL1, an HSPA5 cochaperone. Nat. Genet. 2005, 37, 1309–1311. [Google Scholar] [CrossRef]
- Jauhari, P.; Sahu, J.K.; Roos, A.; Senderek, J.; Vyas, S.; Singhi, P. SIL1-negative Marinesco-Sjögren syndrome: First report of two sibs from India. J. Pediatr. Neurosci. 2014, 9, 291–292. [Google Scholar]
- Senderek, J.; Krieger, M.; Stendel, C.; Bergmann, C.; Moser, M.; Breitbach-Faller, N.; Rudnik-Schöneborn, S.; Blaschek, A.; Wolf, N.; Harting, I.; et al. Mutations in SIL1 cause Marinesco-Sjögren syndrome, a cerebellar ataxia with cataract and myopathy. Nat. Genet. 2005, 37, 1312–1314. [Google Scholar] [CrossRef]
- Buchkremer, S.; Coraspe, J.A.G.; Weis, J.; Roos, A. Sil1-Mutant Mice Elucidate Chaperone Function in Neurological Disorders. J. Neuromuscul. Dis. 2016, 3, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Rosales, C.; Seburn, K.; Ron, D.; Ackerman, S.L. Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjögren syndrome. Hum. Mol. Genet. 2010, 19, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Delépine, M.; Nicolino, M.; Barrett, T.; Golamaully, M.; Lathrop, G.M.; Julier, C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Blumen, S.C.; Astord, S.; Robin, V.; Vignaud, L.; Toumi, N.; Cieslik, A.; Achiron, A.; Carasso, R.L.; Gurevich, M.; Braverman, I.; et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann. Neurol. 2012, 71, 509–519. [Google Scholar] [CrossRef]
- Khanim, F.; Kirk, J.; Latif, F.; Barrett, T.G. WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum. Mutat. 2001, 17, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Haack, T.B.; Kopajtich, R.; Gorza, M.; Rapaport, D.; Greiner, M.; Schönfeld, C.; Freiberg, C.; Schorr, S.; Holl, R.W.; et al. Absence of BiP Co-chaperone DNAJC3 Causes Diabetes Mellitus and Multisystemic Neurodegeneration. Am. J. Hum. Genet. 2014, 95, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Bublitz, S.K.; Alhaddad, B.; Synofzik, M.; Kuhl, V.; Lindner, A.; Freiberg, C.; Schmidt, H.; Strom, T.; Haack, T.; Deschauer, M. Expanding the phenotype of DNAJC3 mutations: A case with hypothyroidism additionally to diabetes mellitus and multisystemic neurodegeneration. Clin. Genet. 2017, 92, 561–562. [Google Scholar] [CrossRef]
- Lytrivi, M.; Senée, V.; Salpea, P.; Fantuzzi, F.; Philippi, A.; Abdulkarim, B.; Sawatani, T.; Marín-Cañas, S.; Pachera, N.; Degavre, A.; et al. DNAJC3 deficiency induces β-cell mitochondrial apoptosis and causes syndromic young-onset diabetes. Eur. J. Endocrinol. 2021, 184, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.-X.; Cao, D.-L.; Xiong, Y.; Peng, X.-H.; Liao, D.-F. A novel model of cholesterol efflux from lipid-loaded cells. Acta Pharmacol. Sin. 2010, 31, 1243–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Shyu, P.; Ng, B.S.H.; Ho, N.; Chaw, R.; Seah, Y.L.; Marvalim, C.; Thibault, G. Membrane phospholipid alteration causes chronic ER stress through early degradation of homeostatic ER-resident proteins. Sci. Rep. 2019, 9, 8637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volmer, R.; Ron, D. Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.; Biden, T.J. Alteration of Endoplasmic Reticulum Lipid Rafts Contributes to Lipotoxicity in Pancreatic β-Cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Taubert, S. Beyond Proteostasis: Lipid Metabolism as a New Player in ER Homeostasis. Metabolites 2021, 11, 52. [Google Scholar] [CrossRef]
- Izawa, T.; Tsuboi, T.; Kuroha, K.; Inada, T.; Nishikawa, S.-I.; Endo, T. Roles of Dom34:Hbs1 in Nonstop Protein Clearance from Translocators for Normal Organelle Protein Influx. Cell Rep. 2012, 2, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, S.; Yunoki, K.; Izawa, T.; Tamura, Y.; Nishikawa, S.-I.; Endo, T. Quality control of nonstop membrane proteins at the ER membrane and in the cytosol. Sci. Rep. 2016, 6, 30795. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, K.; Zhang, Y.; Princen, K.; Hatse, S.; Samala, M.F.; Dey, K.; Choi, H.-J.; Ahn, Y.; Sodoma, A.; Snoeck, R.; et al. CADA Inhibits Human Immunodeficiency Virus and Human Herpesvirus 7 Replication by Down-modulation of the Cellular CD4 Receptor. Virology 2002, 302, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, K.; Schols, D. Cyclotriazadisulfonamides: Promising new CD4-targeted anti-HIV drugs. J. Antimicrob. Chemother. 2005, 56, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, K.; Bell, T.W.; Van Puyenbroeck, V.; Giraut, A.; Noppen, S.; Liekens, S.; Schols, D.; Hartmann, E.; Kalies, K.-U.; Marsh, M. Signal Peptide-Binding Drug as a Selective Inhibitor of Co-Translational Protein Translocation. PLoS Biol. 2014, 12, e1002011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeire, K.; Princen, K.; Hatse, S.; De Clercq, E.; Dey, K.; Bell, T.W.; Schols, D. CADA, a novel CD4-targeted HIV inhibitor, is synergistic with various anti-HIV drugs in vitro. AIDS 2004, 18, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Van Puyenbroeck, V.; Pauwels, E.; Provinciael, B.; Bell, T.W.; Schols, D.; Kalies, K.-U.; Hartmann, E.; Vermeire, K. Preprotein signature for full susceptibility to the co-translational translocation inhibitor cyclotriazadisulfonamide. Traffic 2020, 21, 250–264. [Google Scholar] [CrossRef]
- Van Puyenbroeck, V.; Claeys, E.; Schols, D.; Bell, T.W.; Vermeire, K. A Proteomic Survey Indicates Sortilin as a Secondary Substrate of the ER Translocation Inhibitor Cyclotriazadisulfonamide (CADA). Mol. Cell. Proteom. 2017, 16, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Claeys, E.; Pauwels, E.; Humblet-Baron, S.; Provinciael, B.; Schols, D.; Waer, M.; Sprangers, B.; Vermeire, K. Small Molecule Cyclotriazadisulfonamide Abrogates the Upregulation of the Human Receptors CD4 and 4-1BB and Suppresses In Vitro Activation and Proliferation of T Lymphocytes. Front. Immunol. 2021, 12, 1340. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, E.; Rutz, C.; Provinciael, B.; Stroobants, J.; Schols, D.; Hartmann, E.; Krause, E.; Stephanowitz, H.; Schülein, R.; Vermeire, K. A Proteomic Study on the Membrane Protein Fraction of T Cells Confirms High Substrate Selectivity for the ER Translocation Inhibitor Cyclotriazadisulfonamide. Mol. Cell. Proteom. 2021, 20, 100144. [Google Scholar] [CrossRef]
- Pauwels, E.; Schülein, R.; Vermeire, K. Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway. Int. J. Mol. Sci. 2021, 22, 12007. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. Dual topology of co-chaperones at the membrane of the endoplasmic reticulum. Cell Death Discov. 2021, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Mitra, D.; Salerno, J.R.; Hegde, R.S. Signal Sequences Control Gating of the Protein Translocation Channel in a Substrate-Specific Manner. Dev. Cell 2002, 2, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Fernández, A.; Llacuna, L.; Fernández-Checa, J.C.; Colell, A. Mitochondrial Cholesterol Loading Exacerbates Amyloid β Peptide-Induced Inflammation and Neurotoxicity. J. Neurosci. 2009, 29, 6394–6405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquer, C.; Laine, J.; Dauphinot, L.; Hanbouch, L.; Lemercier-Neuillet, C.; Pierrot, N.; Bossers, K.; Le, M.; Corlier, F.; Benstaali, C.; et al. Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol. Neurodegener. 2014, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Pelchen-Matthews, A.; Boulet, I.; Littman, D.R.; Fagard, R.; Marsh, M. The protein tyrosine kinase p56lck inhibits CD4 endocytosis by preventing entry of CD4 into coated pits. J. Cell Biol. 1992, 117, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.W.; Anugu, S.; Bailey, P.; Catalano, V.J.; Dey, K.; Drew, M.G.B.; Duffy, N.H.; Jin, Q.; Samala, M.F.; Sodoma, A.; et al. Synthesis and Structure−Activity Relationship Studies of CD4 Down-Modulating Cyclotriazadisulfonamide (CADA) Analogues. J. Med. Chem. 2006, 49, 1291–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauwels, E.; Provinciael, B.; Camps, A.; Hartmann, E.; Vermeire, K. Reduced DNAJC3 Expression Affects Protein Translocation across the ER Membrane and Attenuates the Down-Modulating Effect of the Translocation Inhibitor Cyclotriazadisulfonamide. Int. J. Mol. Sci. 2022, 23, 584. https://doi.org/10.3390/ijms23020584
Pauwels E, Provinciael B, Camps A, Hartmann E, Vermeire K. Reduced DNAJC3 Expression Affects Protein Translocation across the ER Membrane and Attenuates the Down-Modulating Effect of the Translocation Inhibitor Cyclotriazadisulfonamide. International Journal of Molecular Sciences. 2022; 23(2):584. https://doi.org/10.3390/ijms23020584
Chicago/Turabian StylePauwels, Eva, Becky Provinciael, Anita Camps, Enno Hartmann, and Kurt Vermeire. 2022. "Reduced DNAJC3 Expression Affects Protein Translocation across the ER Membrane and Attenuates the Down-Modulating Effect of the Translocation Inhibitor Cyclotriazadisulfonamide" International Journal of Molecular Sciences 23, no. 2: 584. https://doi.org/10.3390/ijms23020584
APA StylePauwels, E., Provinciael, B., Camps, A., Hartmann, E., & Vermeire, K. (2022). Reduced DNAJC3 Expression Affects Protein Translocation across the ER Membrane and Attenuates the Down-Modulating Effect of the Translocation Inhibitor Cyclotriazadisulfonamide. International Journal of Molecular Sciences, 23(2), 584. https://doi.org/10.3390/ijms23020584