
Solvent-Free Formation of Cyclodextrin-Based Pseudopolyrotaxanes of Polyethylene Glycol: Kinetic and Structural Aspects




Abstract
:1. Introduction
2. Results and Discussion
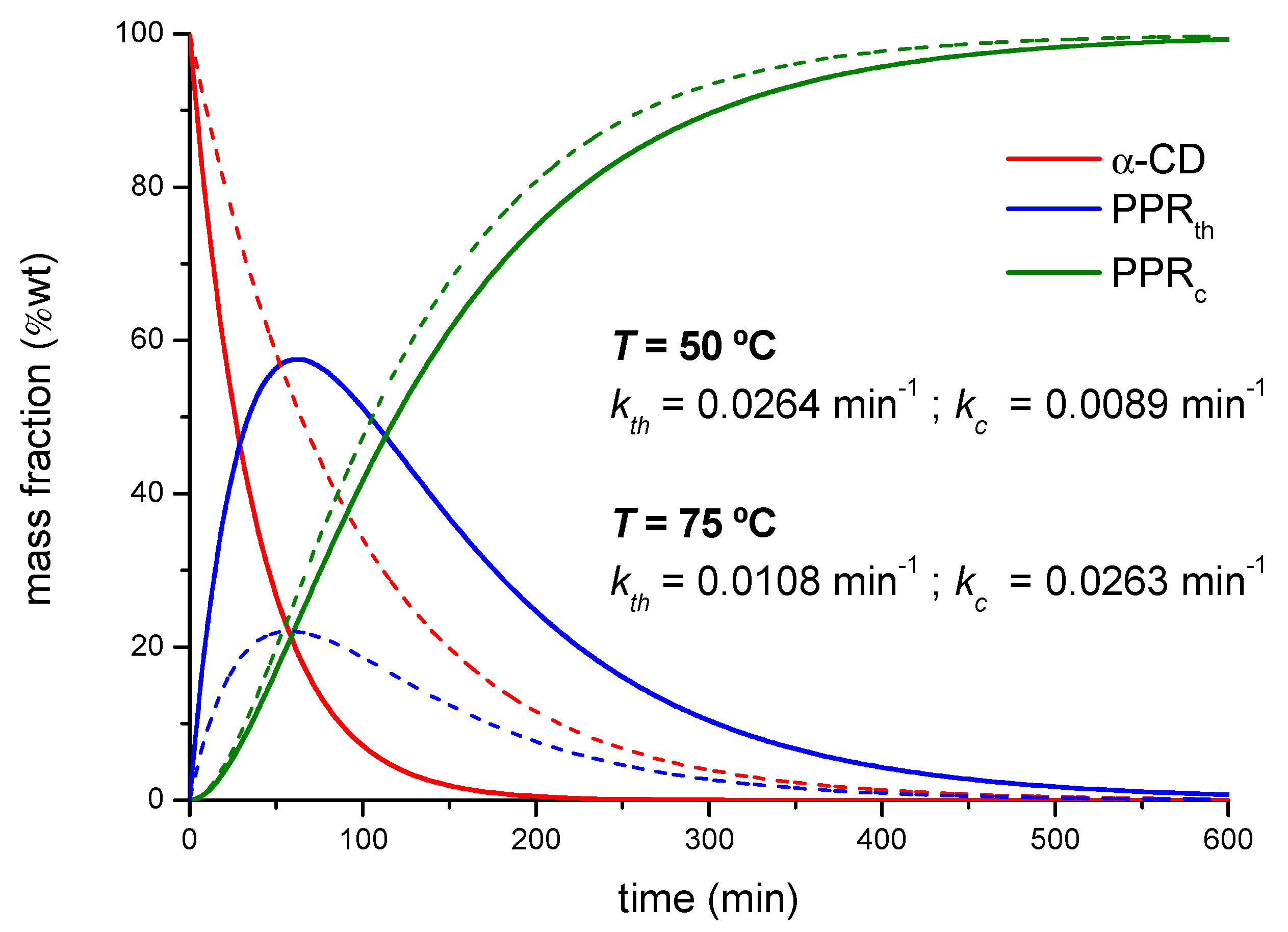
2.1. Mechanism of the PPR Formation
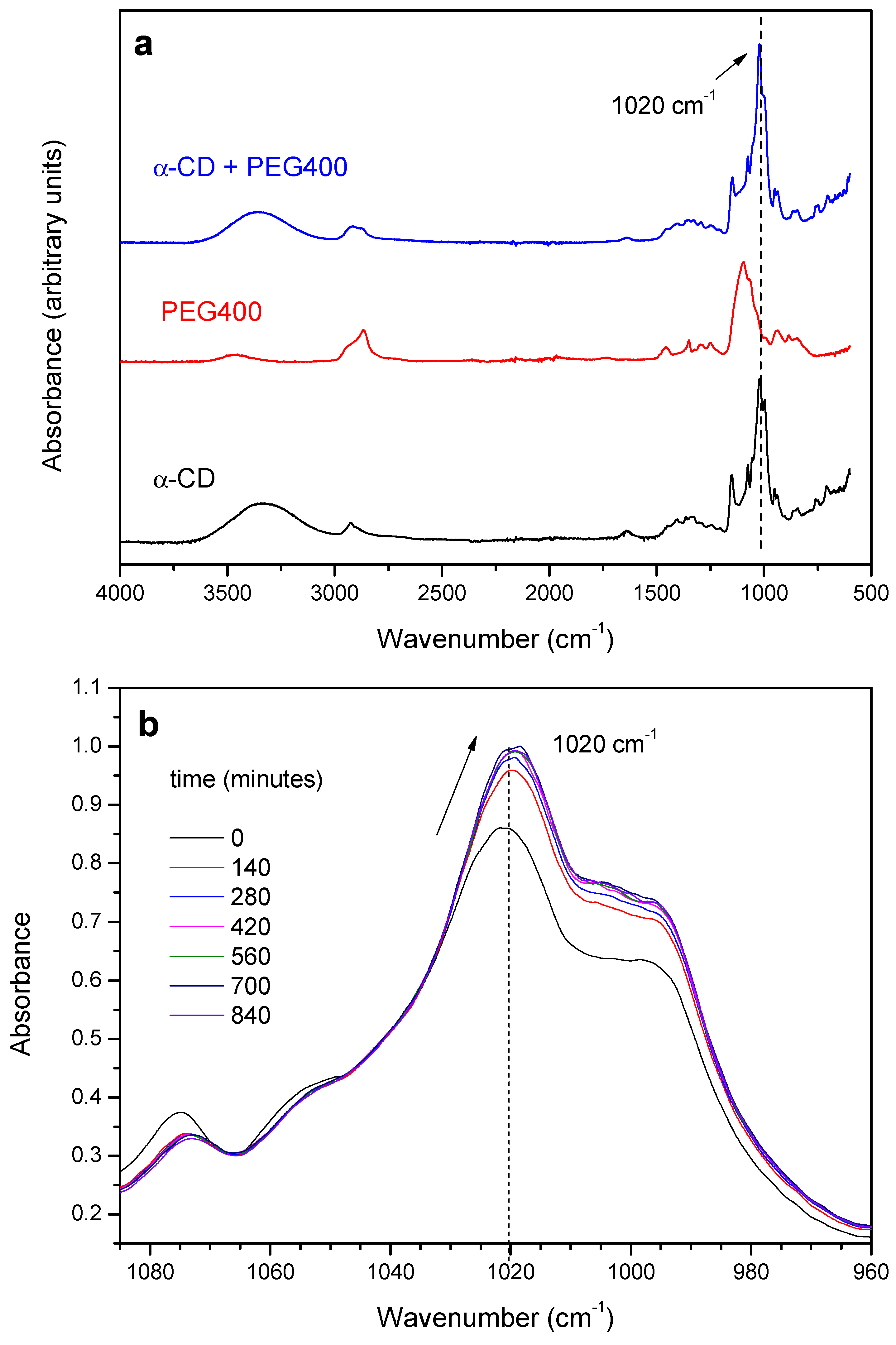
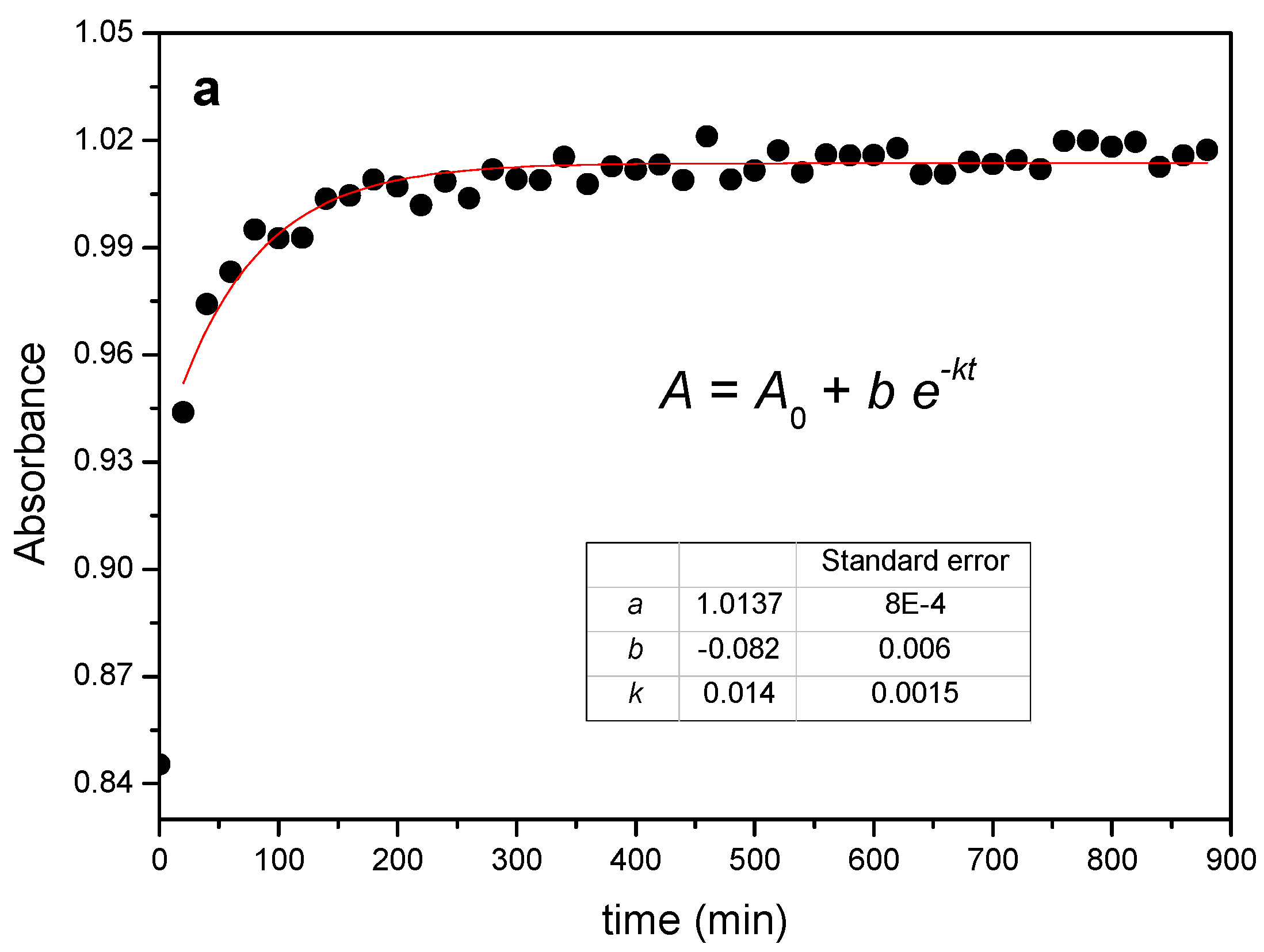
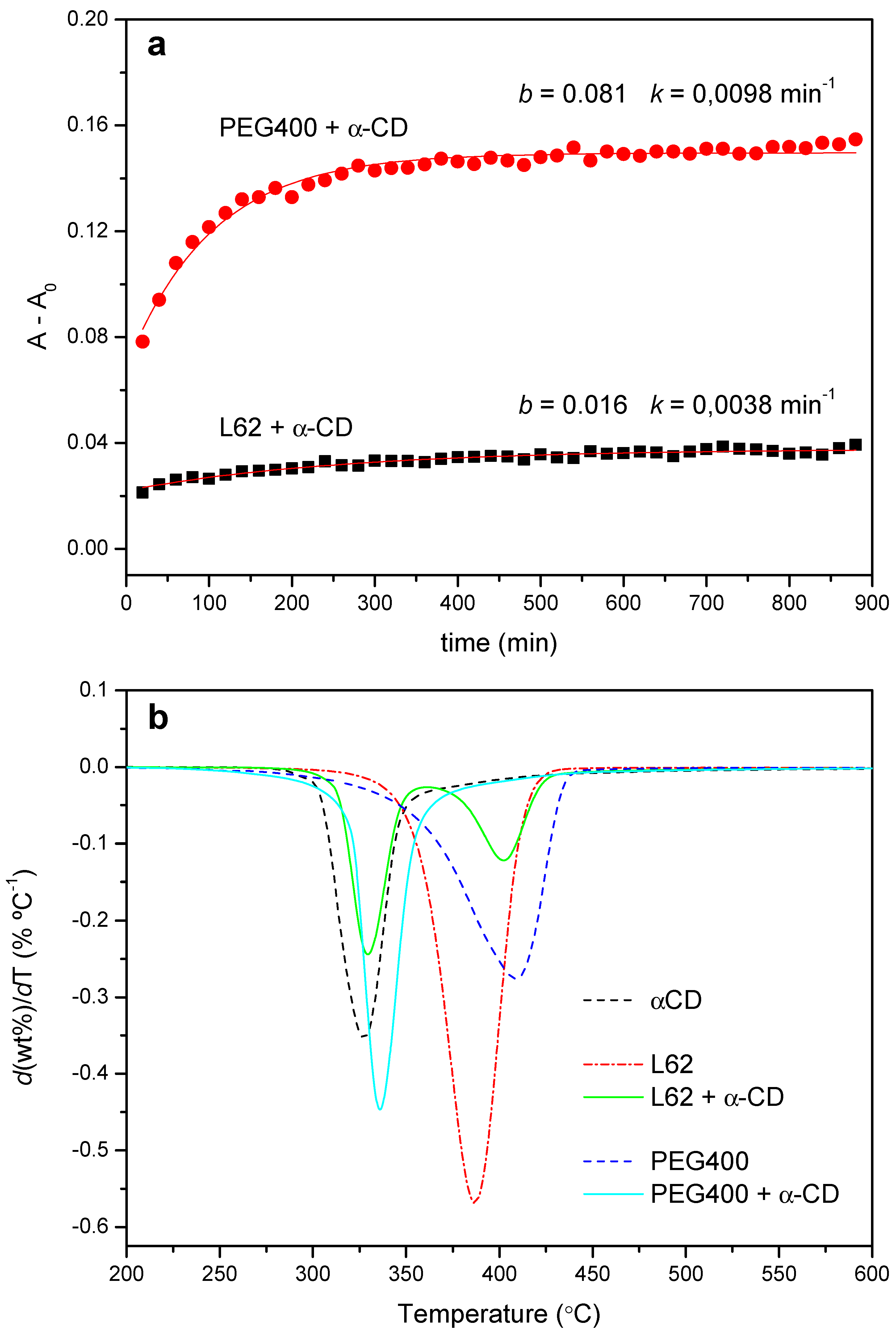
2.1.1. Threading Step (Formation of the PPR)
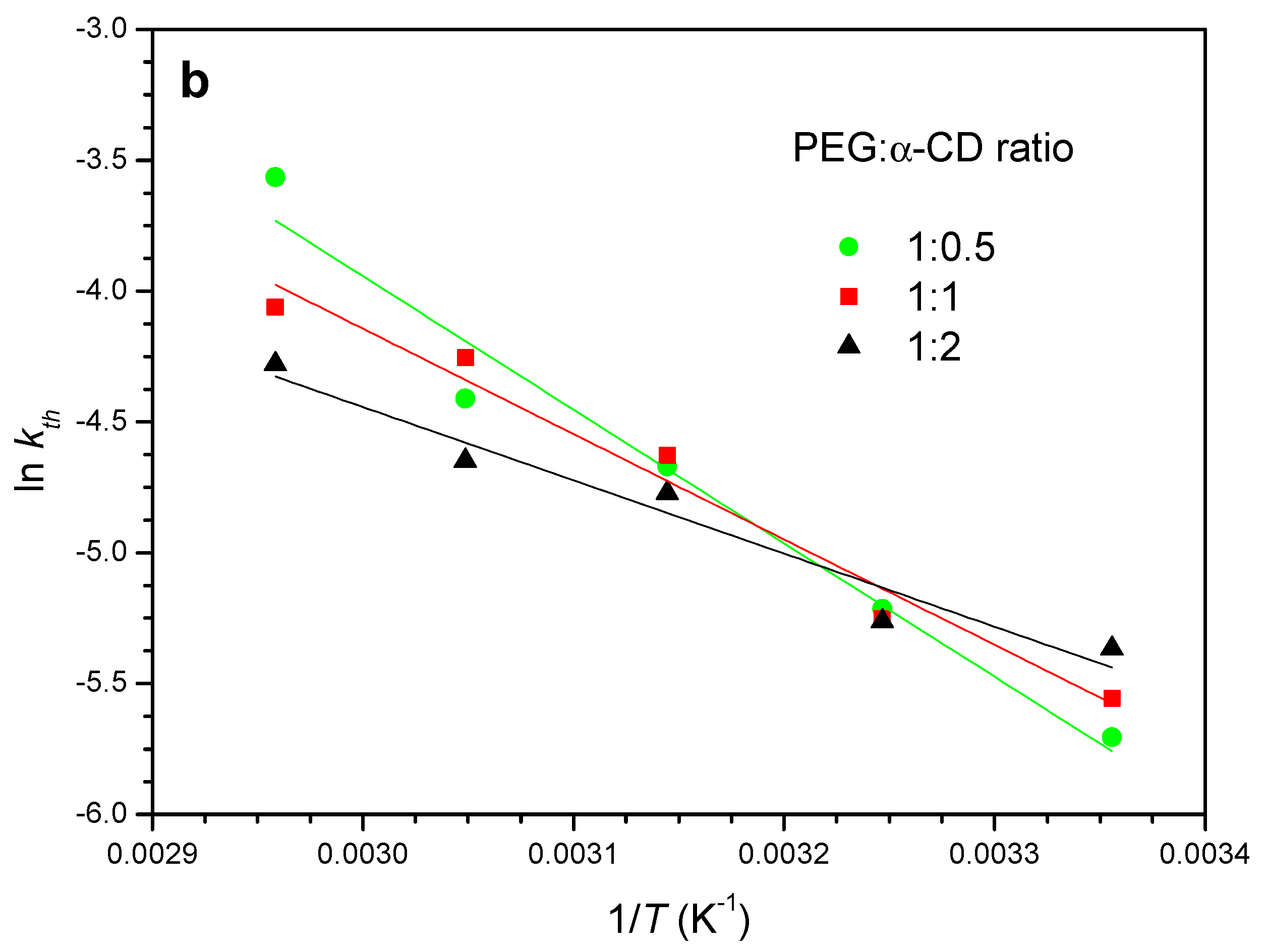
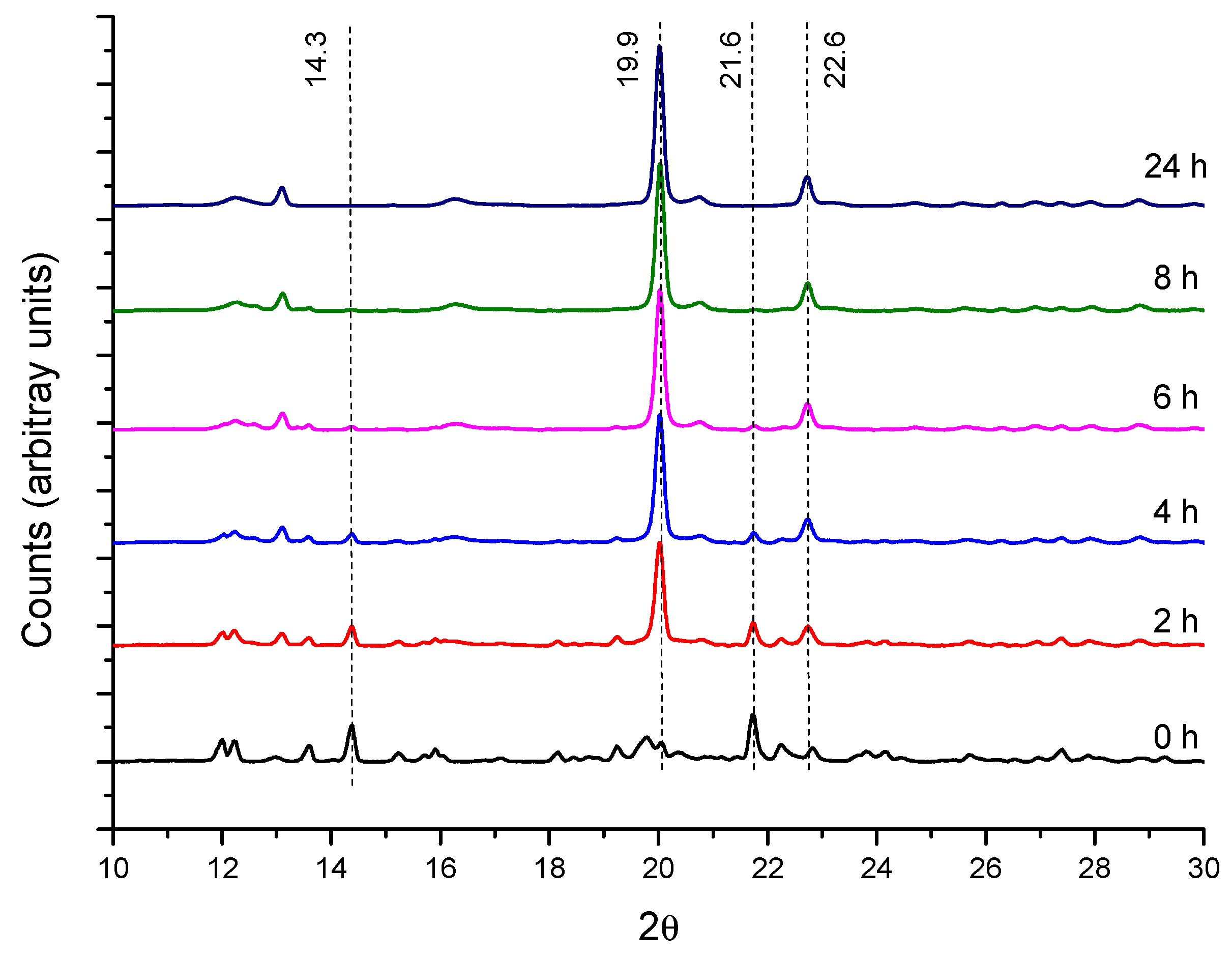
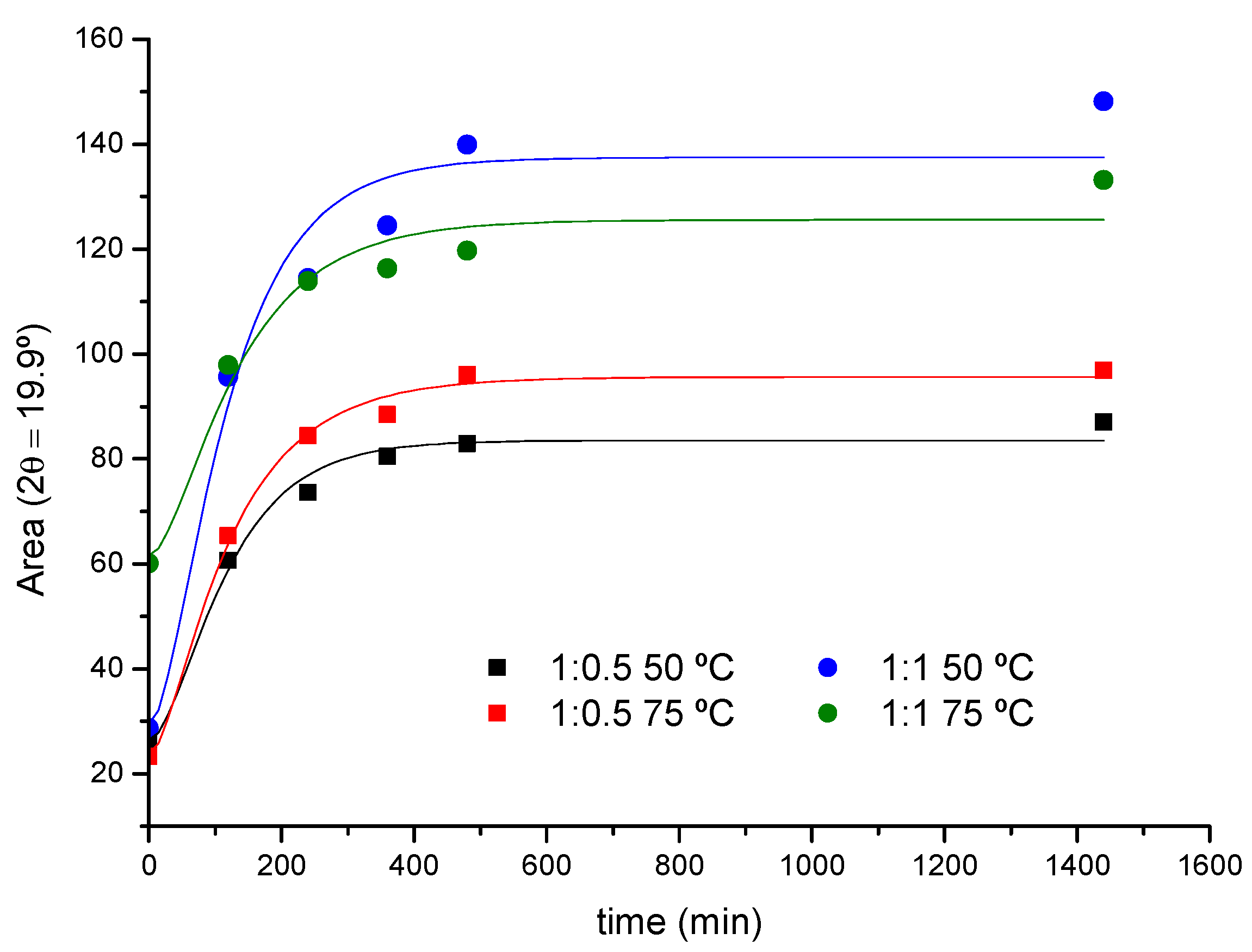
2.1.2. Crystallization Step (Formation of Crystalline PPR)
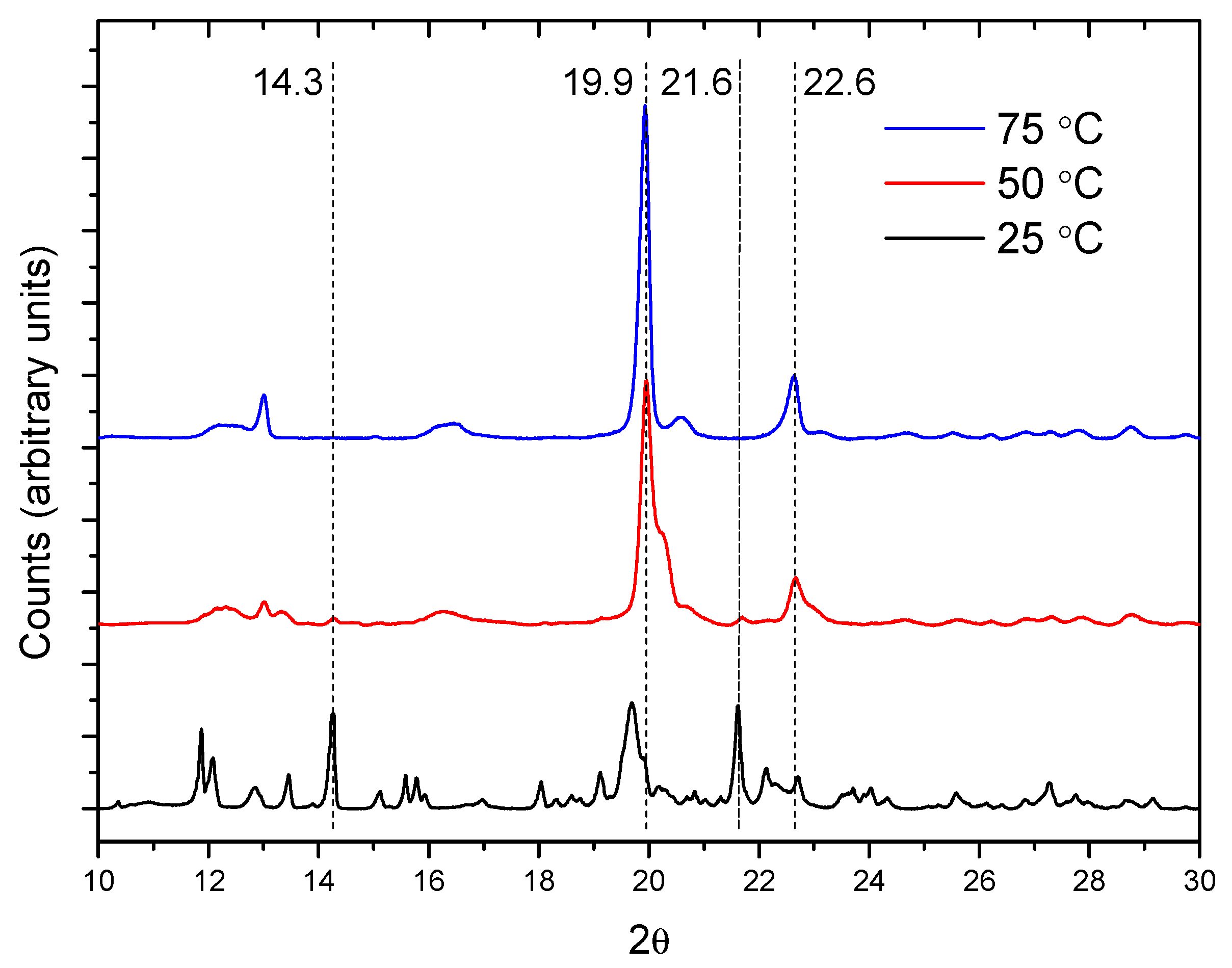
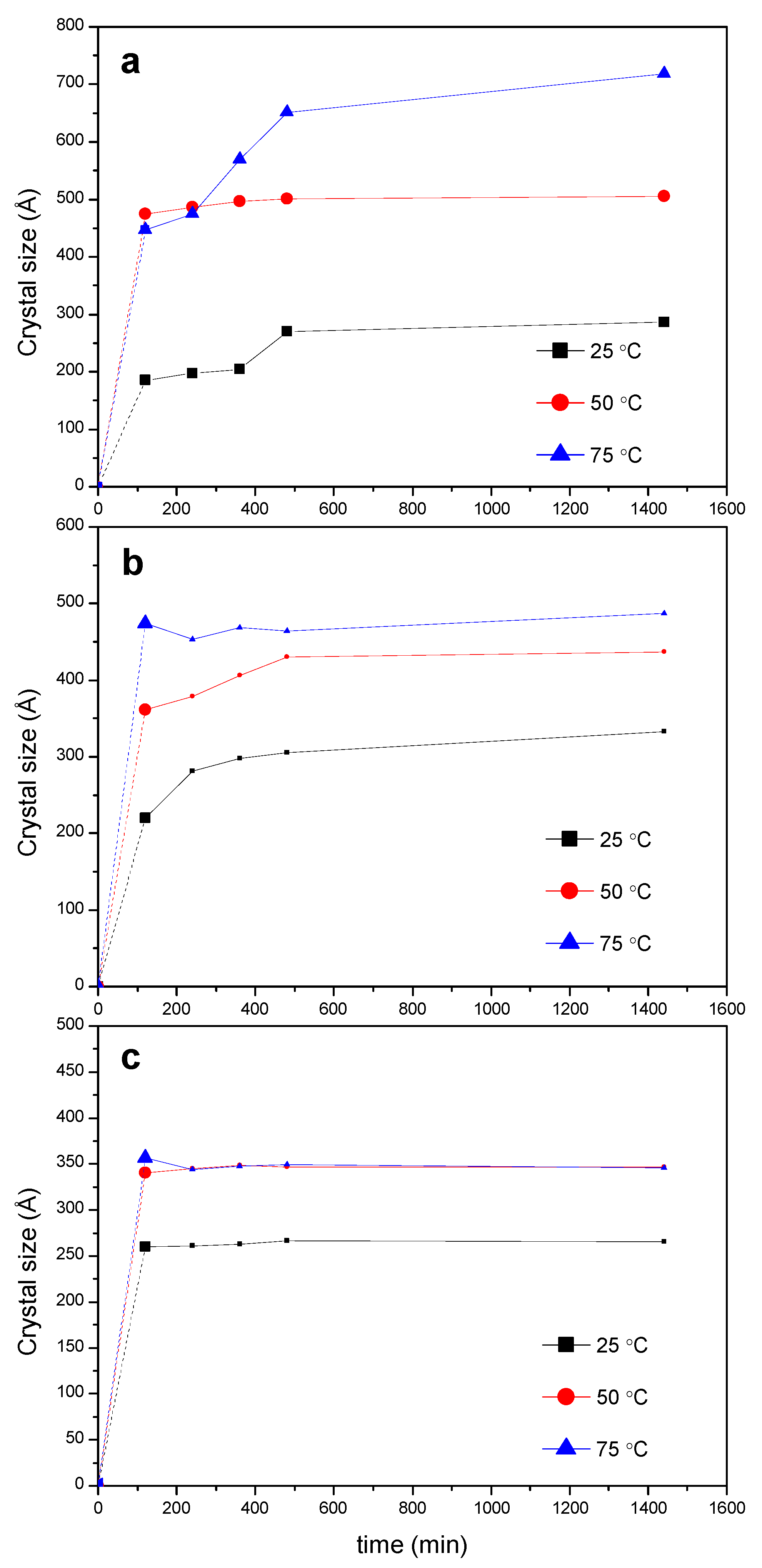
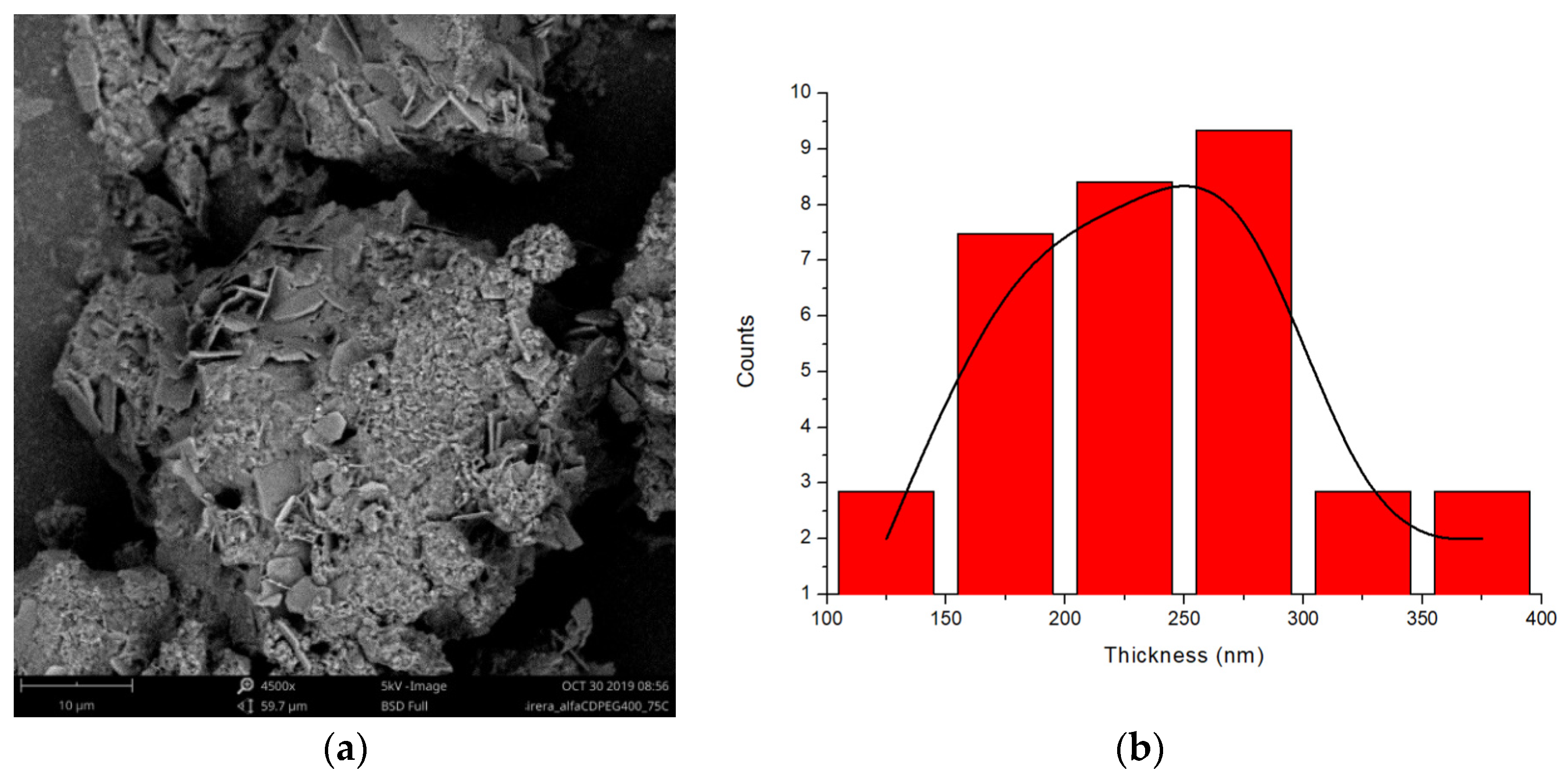
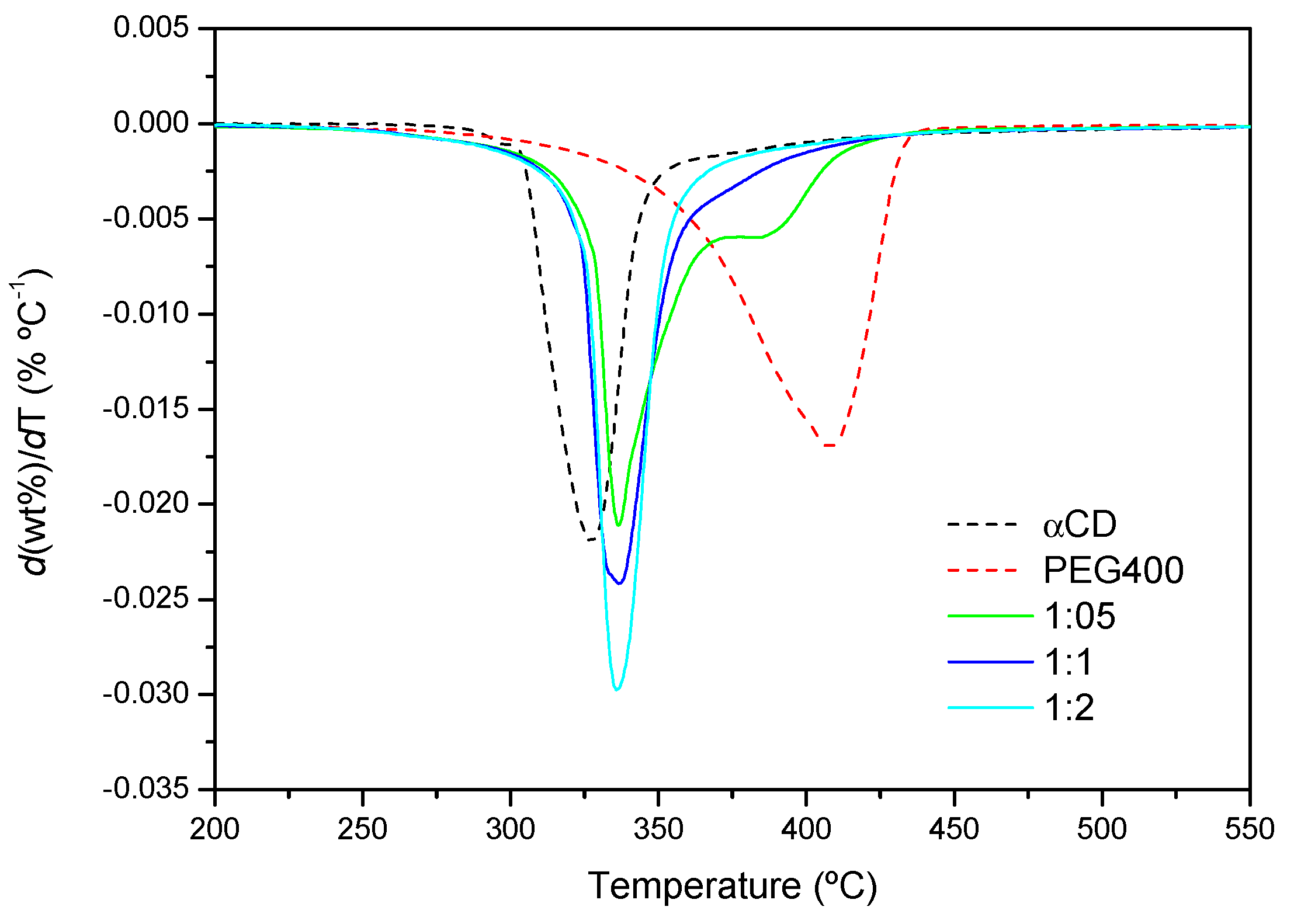
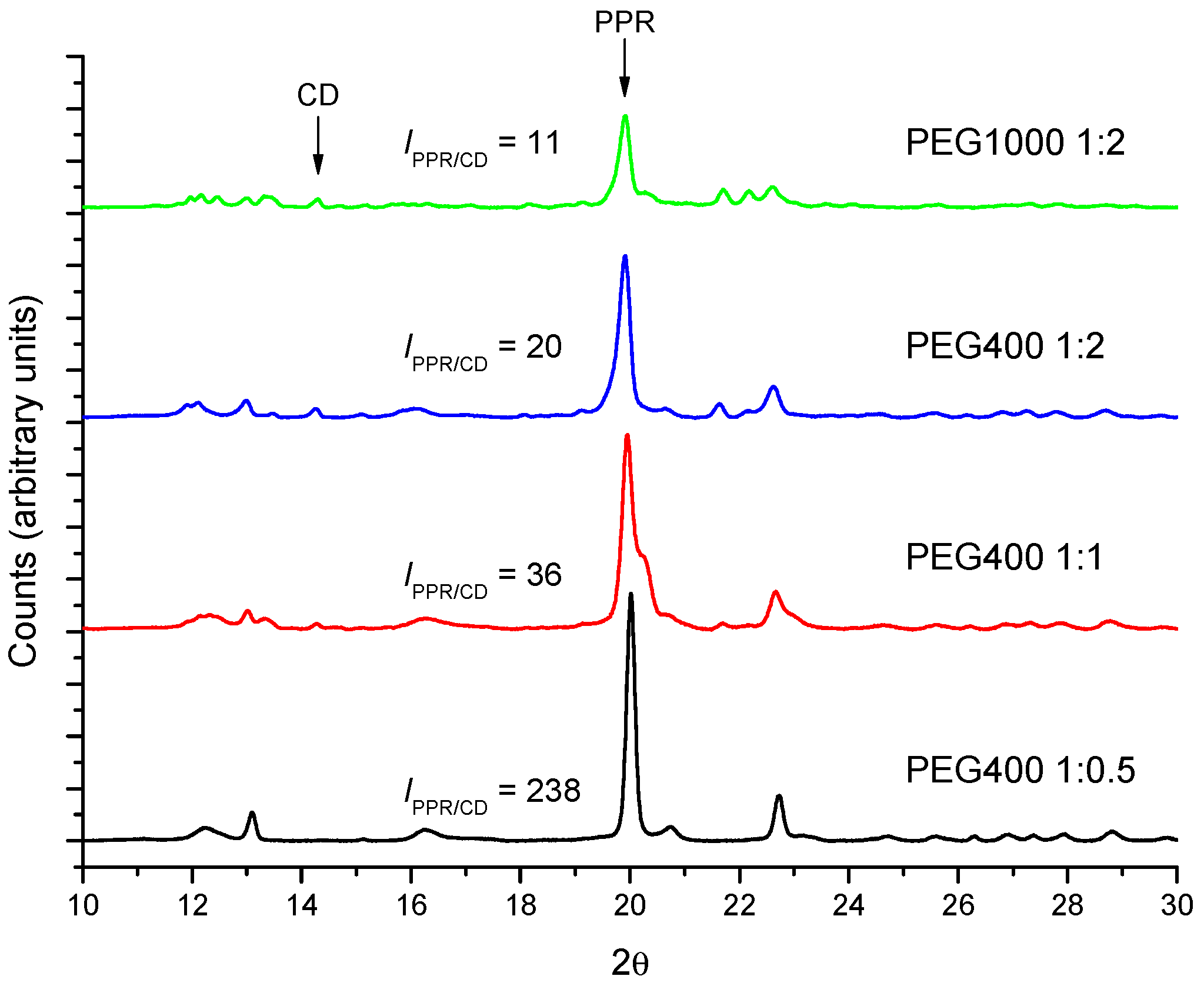
2.2. PPR Structure and Stability
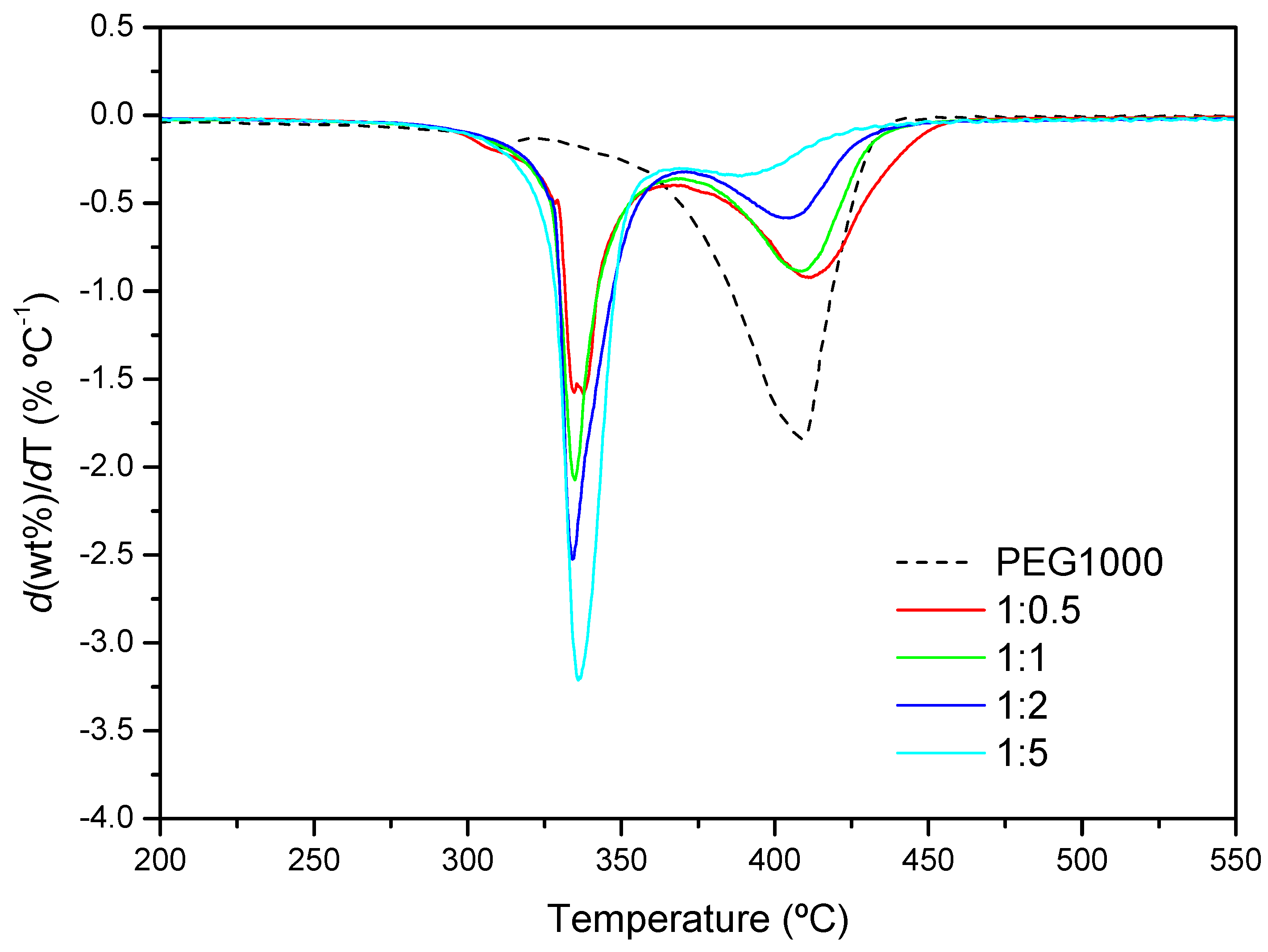
2.3. Effects of the Polymer Chain Length and Structure
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of the Pseudopolyrotaxanes
3.3. FTIR-ATR Characterization and Kinetics
3.4. XRD Characterization
3.5. Derivative Thermogravimetric Analysis
3.6. Scanning Electron Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harada, A.; Li, J.; Kamachi, M. The Molecular Necklace: A Rotaxane Containing Many Threaded α-Cyclodextrins. Nature 1992, 356, 325–327. [Google Scholar] [CrossRef]
- Wenz, G. Cyclodextrins as Building Blocks for Supramolecular Structures and Functional Units. Angew. Chem. Int. Ed. Engl. 1994, 33, 803–822. [Google Scholar] [CrossRef]
- Nepogodiev, S.A.; Stoddart, J.F. Cyclodextrin-Based Catenanes and Rotaxanes. Chem. Rev. 1998, 98, 1959–1976. [Google Scholar] [CrossRef] [PubMed]
- Egele, K.; Samaddar, S.; Schneider, N.; Thompson, D.; Wenz, G. Synthesis of the anionic Hydroxypropyl-β-Cyclodextrin:Poly(decamethylenephosphate) polyrotaxane and Evaluation of its Cholesterol Efflux Potential in Niemann-Pick C1 Cells. J. Mater. Chem. B 2019, 7, 528–537. [Google Scholar] [CrossRef]
- Bai, S.; Hou, M.; Shi, X.; Chen, J.; Ma, X.; Gao, Y.; Wang, Y.; Xue, P.; Kang, Y.; Xu, Z. Reduction-Active Polymeric Prodrug Micelles Based on α-cyclodextrin Polyrotaxanes for Triggered Drug Release and Enhanced Cancer Therapy. Carbohydr. Polym. 2018, 193, 153–162. [Google Scholar] [CrossRef]
- Tardy, B.L.; Dam, H.H.; Kamphuis, M.M.J.; Richardson, J.J.; Caruso, F. Self-Assembled Stimuli-Responsive Polyrotaxane Core-Shell Particles. Biomacromolecules 2014, 15, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chipot, C.; Shao, X.; Cai, W. How do α-Cyclodextrins Self-organize on a Polymer Chain? J. Phys. Chem. C. 2012, 116, 17913–17918. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]
- Puig-Rigall, J.; Serra-Gómez, R.; Guembe-Michel, N.; Grillo, I.; Dreiss, C.A.; González-Gaitano, G. Threading Different Rings on X-Shaped Block Copolymers: Hybrid Pseudopolyrotaxanes of Cyclodextrins and Tetronics. Macromolecules 2020, 53, 3166–3174. [Google Scholar] [CrossRef]
- Kamitori, S.; Matsuzaka, O.; Kondo, S.; Muraoka, S.; Okuyama, K.; Noguchi, K.; Okada, M.; Harada, A. Novel Pseudo-Polyrotaxane Structure Composed of Cyclodextrins and a Straight-Chain Polymer: Crystal Structures of Inclusion Complexes of β-Cyclodextrin with Poly(trimethylene Oxide) and Poly(propylene Glycol). Macromolecules 2000, 33, 1500–1502. [Google Scholar] [CrossRef]
- Harada, A. Preparation and Structures of Supramolecules between Cyclodextrins and Polymers. Coord. Chem. Rev. 1996, 148, 115–133. [Google Scholar] [CrossRef]
- Harada, A. Cyclodextrin-based Molecular Machines. Acc. Chem. Res. 2001, 34, 456–464. [Google Scholar] [CrossRef]
- Harada, A.; Kamachi, M. Complex Formation between Poly(ethylene Glycol) and α-Cyclodextrin. Macromolecules 1990, 23, 2821–2823. [Google Scholar] [CrossRef]
- Rusa, C.C.; Luca, C.; Tonelli, A.E. Polymer-Cyclodextrin Inclusion Compounds: Toward New Aspects of Their Inclusion Mechanism. Macromolecules 2001, 34, 1318–1322. [Google Scholar] [CrossRef]
- Tsai, C.C.; Leng, S.; Jeong, K.U.; Van Horn, R.M.; Wang, C.L.; Zhang, W.B.; Graham, M.J.; Huang, J.; Ho, R.M.; Chen, Y.; et al. Supramolecular Structure of β-Cyclodextrin and Poly(ethylene Oxide)-Block-Poly(propylene Oxide)-Block-Poly(ethylene Oxide) Inclusion Complexes. Macromolecules 2010, 43, 9454–9461. [Google Scholar] [CrossRef]
- Yang, C.; Ni, X.; Li, J. Synthesis of Polypseudorotaxanes and Polyrotaxanes with Multiple α- and γ-Cylodextrins Co-Threaded over Poly[ethylene Oxide)-ran-(propylene Oxide)]. Polymer 2009, 50, 4496–4504. [Google Scholar] [CrossRef]
- Ohshita, N.; Motoyama, K.; Iohara, D.; Hirayama, F.; Taharabaru, T.; Watabe, N.; Kawabata, Y.; Onodera, R.; Higashi, T. Polypseudorotaxane-based Supramolecular Hydrogels Consisting of Cyclodextrins and Pluronics as Stabilizing Agents for Antibody Drugs. Carbohydr. Polym. 2021, 256, 117419. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, V.; Cavallaro, G.; Lazzara, G.; Milioto, S.; Parisi, F. Crystallinity of Block Copolymer Controlled by Cyclodextrin. J. Therm. Anal. Calorim. 2018, 132, 191–196. [Google Scholar] [CrossRef]
- Peet, J.; Rusa, C.C.; Hunt, M.A.; Tonelli, A.E.; Balik, C.M. Solid-State Complexation of Poly(ethylene Glycol) with Alpha-Cyclodextrin. Macromolecules 2005, 38, 537–541. [Google Scholar] [CrossRef]
- Douhal, A. Ultrafast guest dynamics in cyclodextrin nanocavities. Chem. Rev. 2004, 104, 1955–1976. [Google Scholar] [CrossRef]
- Harada, A.; Okada, M.; Li, J.; Kamachi, M. Preparation and Characterization of Inclusion Complexes of Poly(propylene glycol) with Cyclodextrins. Macromolecules 1995, 28, 8406–8411. [Google Scholar] [CrossRef]
- Amiel, C.; Sébille, B. Association Between Amphiphilic Poly(ethylene oxide) and β-cyclodextrin Polymers: Aggregation and Phase Separation. Adv. Colloid Interface Sci. 1999, 79, 105–122. [Google Scholar] [CrossRef]
- Choi, S.; Kwon, T.W.; Coskun, A.; Choi, J.W. Highly Elastic Binders Integrating Polyrotaxanes for Silicon Microparticle Anodes in Lithium Ion Batteries. Science 2017, 357, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.Y.; Min, X.; Chao, H.W.; Hai, J.D. Rare Silver Bromide Clustered Supramolecular Polypseudorotaxane: Preparation and Its Application in Photocatalytic Degradation of Organic Compounds. Main Group Chem. 2020, 19, 139–147. [Google Scholar] [CrossRef]
- González-Gaitano, G.; Isasi, J.R.; Vélaz, I.; Zornoza, A. Drug Carrier Systems Based On Cyclodextrin Supramolecular Assemblies And Polymers: Present And Perspectives. Curr. Pharm. Des. 2016, 23, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Puig-Rigall, J.; Serra-Gómez, R.; Stead, I.; Grillo, I.; Dreiss, C.A.; González-Gaitano, G. Pseudo-Polyrotaxanes of Cyclodextrins with Direct and Reverse X-Shaped Block Copolymers: A Kinetic and Structural Study. Macromolecules 2019, 52, 1458–1468. [Google Scholar] [CrossRef] [Green Version]
- Kihara, N.; Hinoue, K.; Takata, T. Solid-State End-Capping of Pseudopolyrotaxane Possessing Hydroxy-Terminated Axle to Polyrotaxane and its Application to the Synthesis of a Functionalized Polyrotaxane Capable of Yielding a Polyrotaxane Network. Macromolecules 2005, 38, 223–226. [Google Scholar] [CrossRef]
- Harada, A.; Okada, M.; Kawaguchi, Y. Formation of Self-Assembled Tubular Structures by Mixing Cyclodextrin and Polymers Without Solvents. Y. Chem. Lett. 2005, 34, 542–543. [Google Scholar] [CrossRef]
- Liu, R.; Harada, A.; Takata, T. Solvent-Free Synthesis of Unmodified Cyclodextrin-Based Pseudopolyrotaxane and Polyrotaxane by Grinding. Polym. J. 2007, 39, 21–23. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Maeda, T.; Kihara, N.; Harada, A.; Takata, T. Solvent-Free Synthesis of Pseudopolyrotaxane and Polyrotaxane: Efficient Threading Complexation of a Cyclodextrin Wheel and a Linear Polymer Axle to Yield Pseudopolyrotaxane and its Fixation to Polyrotaxane by the Direct Grinding of a Solid Mixture. J. Polym. Sci. A1 2007, 45, 1571–1574. [Google Scholar] [CrossRef]
- Girardeau, T.E.; Zhao, T.; Leisen, J.-; Beckham, H.W.; Bucknall, D.G. Solid Inclusion Complexes of α-Cyclodextrin and Perdeuterated Poly(Oxyethylene). Macromolecules 2005, 38, 2261–2270. [Google Scholar] [CrossRef]
- Lazzara, G.; Milioto, S. Copolymer-Cyclodextrin Inclusion Complexes in Water and in the Solid State. A Physico-Chemical Study. J Phys Chem. B. 2008, 112, 11887–11895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavà, C.; Crupi, V.; Guardo, M.; Majolino, D.; Stancanelli, R.; Tommasini, S.; Ventura, C.A.; Venuti, V. Phase solubility and FTIR-ATR studies of idebenone/sulfobutyl ether β-cyclodextrin inclusion complex. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 255–262. [Google Scholar] [CrossRef]
- Stancanelli, R.; Venuti, V.; Arigò, A.; Calabrò, M.L.; Cannavà, C.; Crupi, V.; Majolino, D.; Tommasini, S.; Ventura, C.A. Isoflavone aglycons-sulfobutyl ether-β-cyclodextrin inclusion complexes: In solution and solid state studies. J. Incl. Phenom. Macrocycl. Chem. 2015, 83, 27–36. [Google Scholar] [CrossRef]
- Venuti, V.; Cannavà, C.; Cristiano, M.C.; Fresta, M.; Majolino, D.; Paolino, D.; Stancanelli, R.; Tommasini, S.; Ventura, C.A. A characterization study of resveratrol/sulfobutyl ether-β-cyclodextrin inclusion complex and in vitro anticancer activity. Colloids Surf. B Biointerfaces 2014, 115, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Venuti, V.; Stancanelli, R.; Acri, G.; Crupi, V.; Paladini, G.; Testagrossa, B.; Tommasini, S.; Ventura, C.A.; Majolino, D. “Host-guest” interactions in Captisol®/Coumestrol inclusion complex: UV–vis, FTIR-ATR and Raman studies. J. Mol. Struct. 2017, 1146, 512–521. [Google Scholar] [CrossRef]
- Topchieva, I.N.; Tonelli, A.E.; Panova, I.G.; Matuchina, E.V.; Kalashnikov, F.A.; Gerasimov, V.I.; Rusa, C.C.; Rusa, M.; Hunt, M.A. Two-Phase Channel Structures Based on α-Cyclodextrin−Polyethylene Glycol Inclusion Complexes. Langmuir 2004, 20, 9036–9043. [Google Scholar] [CrossRef]
- OMNICTM Specta Software, 6.0 version; Thermo Nicolet Corporation; Thermo Fisher Scientific: Madison, WI, USA.
- OriginPro, 8.5.0 version; OriginLab Corporation: Northampton, MA, USA.
- Huang, L.; Tonelli, A.E. Polymer Inclusion Compounds. J. Macromol. Sci. Part C Polym. Rev. 1998, 38, 781–837. [Google Scholar] [CrossRef]
- DIFFRAC.EVA, 5.2 version; Bruker Corporation: Billerica, MA, USA.
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (°C) | kth (min−1) | ||
|---|---|---|---|
| 1:0.5 | 1:1 | 1:2 | |
| 25 | (3.3 ± 0.2) × 10−3 | (3.9 ± 0.3) × 10−3 | (4.7 ± 0.4) × 10−3 |
| 35 | (5.4 ± 0.2) × 10−3 | (5.2 ± 0.3) × 10−3 | (5.2 ± 0.4) × 10−3 |
| 45 | (9.4 ± 0.3) × 10−3 | (9.8 ± 0.4) × 10−3 | (8.5 ± 0.5) × 10−3 |
| 55 | (12.1 ± 0.8) × 10−3 | (14 ± 1) × 10−3 | (9.6 ± 0.6) × 10−3 |
| 65 | (28 ± 2) × 10−3 | (17 ± 1) × 10−3 | (13.9 ± 0.9) × 10−3 |
| Ea (kJ mol−1) | 42 ± 4 | 34 ± 4 | 23 ± 3 |
| Molar Ratio | 2θ | kc (min−1) | |
|---|---|---|---|
| 50 °C | 75 °C | ||
| 1:0.5 | 19.9° | (23 ± 7) × 10−3 | (10 ± 2) × 10−3 |
| 1:1 | 19.9° | (26 ± 2) × 10−3 | (9 ± 3) × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guembe-Michel, N.; Durán, A.; Sirera, R.; González-Gaitano, G. Solvent-Free Formation of Cyclodextrin-Based Pseudopolyrotaxanes of Polyethylene Glycol: Kinetic and Structural Aspects. Int. J. Mol. Sci. 2022, 23, 685. https://doi.org/10.3390/ijms23020685
Guembe-Michel N, Durán A, Sirera R, González-Gaitano G. Solvent-Free Formation of Cyclodextrin-Based Pseudopolyrotaxanes of Polyethylene Glycol: Kinetic and Structural Aspects. International Journal of Molecular Sciences. 2022; 23(2):685. https://doi.org/10.3390/ijms23020685
Chicago/Turabian StyleGuembe-Michel, Nerea, Adrián Durán, Rafael Sirera, and Gustavo González-Gaitano. 2022. "Solvent-Free Formation of Cyclodextrin-Based Pseudopolyrotaxanes of Polyethylene Glycol: Kinetic and Structural Aspects" International Journal of Molecular Sciences 23, no. 2: 685. https://doi.org/10.3390/ijms23020685
APA StyleGuembe-Michel, N., Durán, A., Sirera, R., & González-Gaitano, G. (2022). Solvent-Free Formation of Cyclodextrin-Based Pseudopolyrotaxanes of Polyethylene Glycol: Kinetic and Structural Aspects. International Journal of Molecular Sciences, 23(2), 685. https://doi.org/10.3390/ijms23020685