
Kidney Damage Caused by Obesity and Its Feasible Treatment Drugs




Abstract
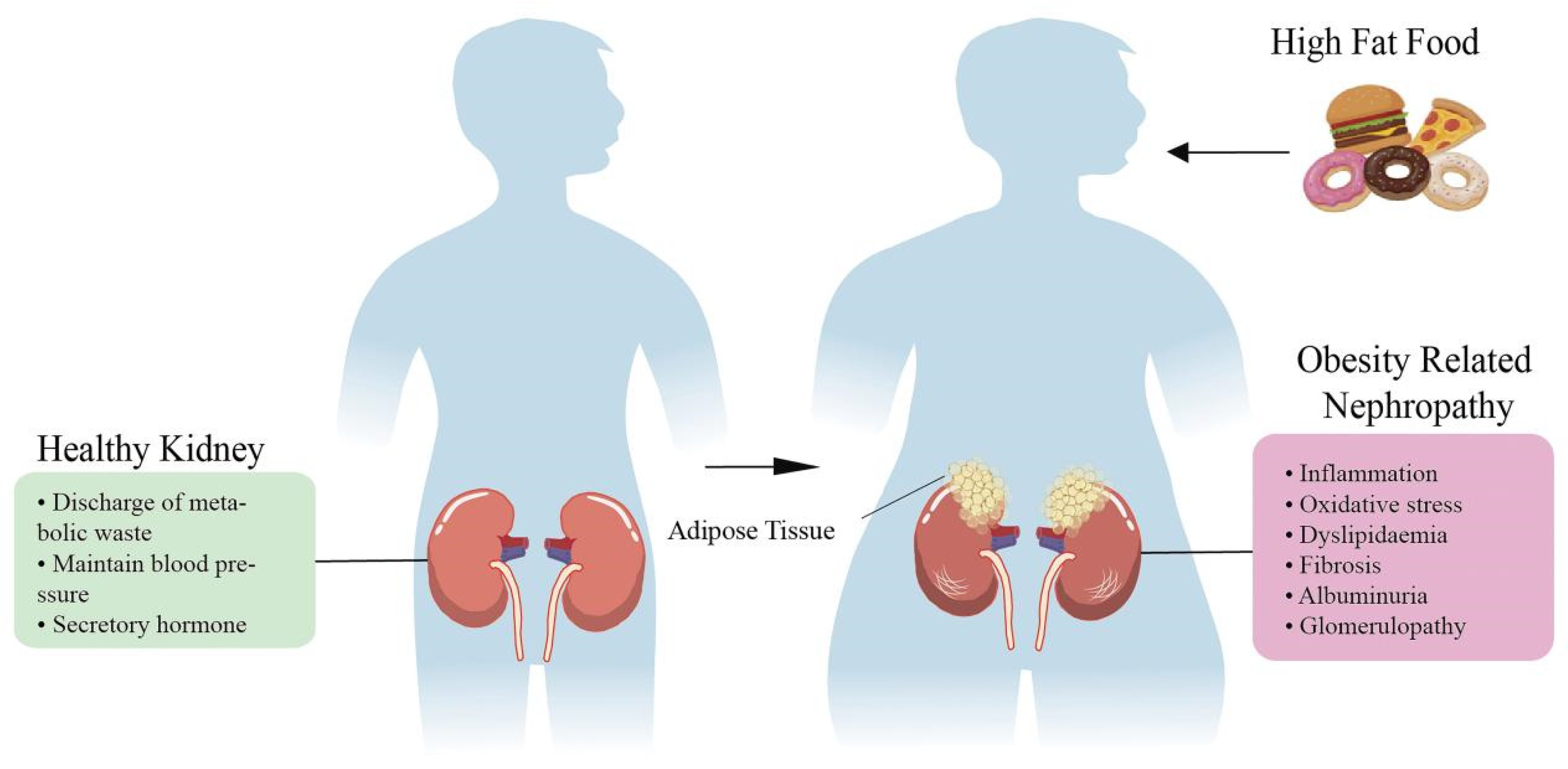
:1. Introduction
2. Obesity-Induced Nephropathy
2.1. Glomerular Injury
2.2. Renal Tubular Injury
3. The Mechanism of Obesity-Induced Nephropathy
3.1. The Inflammatory Response and Insulin Resistance
3.2. Renin–Angiotensin–Aldosterone System
3.3. Lipotoxicity
4. Treatment Drugs for Obesity-Induced Nephropathy
4.1. RAS Inhibitors
4.2. SGLT2 Inhibitors
4.3. Melatonin
5. Perspective
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Min, J.; Zhao, Y.; Slivka, L.; Wang, Y. Double burden of diseases worldwide: Coexistence of undernutrition and overnutrition-related non-communicable chronic diseases. Obes. Rev. 2018, 19, 49–61. [Google Scholar] [CrossRef]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [Green Version]
- James, W.P.T. The epidemiology of obesity: The size of the problem. J. Intern. Med. 2008, 263, 336–352. [Google Scholar] [CrossRef]
- Żukiewicz-Sobczak, W.; Wróblewska, P.; Zwoliński, J.; Chmielewska-Badora, J.; Adamczuk, P.; Krasowska, E.; Zagórski, J.; Oniszczuk, A.; Piątek, J.; Silny, W. Obesity and poverty paradox in developed countries. Ann. Agric. Environ. Med. 2014, 21, 590–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xue, H.; Sun, M.; Zhu, X.; Zhao, L.; Yang, Y. Prevention and control of obesity in China. Lancet Glob. Health 2019, 7, e1166–e1167. [Google Scholar] [CrossRef]
- Gordon-Larsen, P.; Wang, H.; Popkin, B.M. Overweight dynamics in Chinese children and adults. Obes. Rev. 2014, 15 (Suppl. 1), 37–48. [Google Scholar] [CrossRef]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk-Golec, K.; Woźniak, A.; Reiter, R.J. Inter-relationships of the chronobiotic, melatonin, with leptin and adiponectin: Implications for obesity. Pineal Res. 2015, 59, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Meguid El Nahas, A.; Bello, A.K. Chronic kidney disease: The global challenge. Lancet 2005, 365, 331–340. [Google Scholar] [CrossRef]
- Li, P.K.T.; Burdmann, E.A.; Mehta, R.L. Acute kidney injury: Global health alert. Transplantation 2013, 95, 653–657. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Wang, H.; Zhao, X.; Matsushita, K.; Coresh, J.; Zhang, L.; Zhao, M.-H. CKD in China: Evolving spectrum and public health implications. Am. J. Kidney Dis. 2020, 76, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; McMacken, M.; Kalantar-Zadeh, K. Plant-based diets for kidney disease: A guide for clinicians. Am. J. Kidney Dis. 2021, 77, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Preble, W. Obesity: Observations on one thousand cases. Boston Med. Surg. J. 1923, 188, 617–621. [Google Scholar] [CrossRef]
- Hsu, C.-Y.; McCulloch, C.E.; Iribarren, C.; Darbinian, J.; Go, A.S. Body mass index and risk for end-stage renal disease. Ann. Intern. Med. 2006, 144, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, S.; Esmaeili, H.; Alizadeh, M.; Daneshzad, E.; Sharifi, L.; Radfar, H.; Radaei, M.K. Metabolic phenotypes of obese, overweight, and normal weight individuals and risk of chronic kidney disease: A systematic review and meta-analysis. Arch. Endocrinol. Metab. 2019, 63, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Berthoux, F.; Mariat, C.; Maillard, N. Overweight/obesity revisited as a predictive risk factor in primary IgA nephropathy. Nephrol. Dial. Transplant. 2013, 28 (Suppl. 4), iv160–iv166. [Google Scholar] [CrossRef]
- Curran, S.P.; Famure, O.; Li, Y.; Kim, S.J. Increased recipient body mass index is associated with acute rejection and other adverse outcomes after kidney transplantation. Transplantation 2014, 97, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.-L.; Amann, K.; Ritz, E. Nephron number and renal risk in hypertension and diabetes. J. Am. Soc. Nephrol. 2005, 16 (Suppl. 1), S27–S29. [Google Scholar] [PubMed]
- Tsuboi, N.; Koike, K.; Hirano, K.; Utsunomiya, Y.; Kawamura, T.; Hosoya, T. Clinical features and long-term renal outcomes of Japanese patients with obesity-related glomerulopathy. Clin. Exp. Nephrol. 2013, 17, 379–385. [Google Scholar] [CrossRef]
- Okabayashi, Y.; Tsuboi, N.; Sasaki, T.; Haruhara, K.; Kanzaki, G.; Koike, K.; Shimizu, A.; D’Agati, V.D.; Yokoo, T. Single-nephron GFR in patients with obesity-related glomerulopathy. Kidney Int. Rep. 2020, 5, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.D.; Chagnac, A.; de Vries, A.P.J.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef]
- Okabayashi, Y.; Tsuboi, N.; Sasaki, T.; Haruhara, K.; Kanzaki, G.; Koike, K.; Miyazaki, Y.; Kawamura, T.; Ogura, M.; Yokoo, T. Glomerulopathy associated with moderate obesity. Kidney Int. Rep. 2016, 1, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levey, A.S.; Becker, C.; Inker, L.A. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: A systematic review. JAMA 2015, 313, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Tobar, A.; Ori, Y.; Benchetrit, S.; Milo, G.; Herman-Edelstein, M.; Zingerman, B.; Lev, N.; Gafter, U.; Chagnac, A. Proximal tubular hypertrophy and enlarged glomerular and proximal tubular urinary space in obese subjects with proteinuria. PLoS ONE 2013, 8, e75547. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Long, J.; Jiang, W.; Shi, Y.; He, X.; Zhou, Z.; Li, Y.; Yeung, R.O.; Wang, J.; Matsushita, K.; et al. Trends in chronic kidney disease in China. N. Engl. J. Med. 2016, 375, 905–906. [Google Scholar] [CrossRef] [PubMed]
- Kambham, N.; Markowitz, G.S.; Valeri, A.M.; Lin, J.; D’Agati, V.D. Obesity-related glomerulopathy: An emerging epidemic. Kidney Int. 2001, 59, 1498–1509. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, N.; Utsunomiya, Y.; Kanzaki, G.; Koike, K.; Ikegami, M.; Kawamura, T.; Hosoya, T. Low glomerular density with glomerulomegaly in obesity-related glomerulopathy. Clin. J. Am. Soc. Nephrol. 2012, 7, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-M.; Liu, Z.-H.; Zeng, C.-H.; Li, S.-J.; Wang, Q.-W.; Li, L.-S. Podocyte lesions in patients with obesity-related glomerulopathy. Am. J. Kidney Dis. 2006, 48, 772–779. [Google Scholar] [CrossRef]
- Hodgin, J.B.; Bitzer, M.; Wickman, L.; Afshinnia, F.; Wang, S.Q.; O’Connor, C.; Yang, Y.; Meadowbrooke, C.; Chowdhury, M.; Kikuchi, M.; et al. Glomerular aging and focal global glomerulosclerosis: A podometric perspective. J. Am. Soc. Nephrol. 2015, 26, 3162–3178. [Google Scholar] [CrossRef]
- Chen, H.-M.; Li, S.-J.; Chen, H.-P.; Wang, Q.-W.; Li, L.-S.; Liu, Z.-H. Obesity-related glomerulopathy in China: A case series of 90 patients. Am. J. Kidney Dis. 2008, 52, 58–65. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Renal function in diabetic disease models: The tubular system in the pathophysiology of the diabetic kidney. Annu. Rev. Physiol. 2012, 74, 351–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declèves, A.-E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayam, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMK-activated kinase in high fat diet-induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.J.X.; Stock, E.; Broeckx, B.J.G.; Daminet, S.; Meyer, E.; Delanghe, J.R.; Croubels, S.; Devreese, M.; Nguyen, P.; Bogaerts, E.; et al. Weight-gain induced changes in renal perfusion assessed by contrast-enhanced ultrasound precede increases in urinary protein excretion suggestive of glomerular and tubular injury and normalize after weight-loss in dogs. PLoS ONE 2020, 15, e0231662. [Google Scholar]
- Qiao, Y.-F.; Guo, W.-J.; Li, L.; Shao, S.; Qiao, X.; Shao, J.-J.; Zhang, Q.; Li, R.-S.; Wang, L.-H. Melatonin attenuates hypertension-induced renal injury partially through inhibiting oxidative stress in rats. Mol. Med. Rep. 2016, 13, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Nespoux, J.; Vallon, V. SGLT2 inhibition and kidney protection. Clin. Sci. 2018, 132, 1329–1339. [Google Scholar] [CrossRef]
- Novikov, A.; Vallon, V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: An update. Curr. Opin. Nephrol. Hypertens. 2016, 25, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Alicic, R.Z.; Johnson, E.J.; Tuttle, K.R. SGLT2 Inhibition for the Prevention and Treatment of Diabetic Kidney Disease: A Review. Am. J. Kidney Dis. 2018, 72, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Choi, M.-S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. Pract. 2005, 115, 911–919. [Google Scholar] [CrossRef]
- Villarroya, F.; Cereijo, R.; Gavaldà-Navarro, A.; Villarroya, J.; Giralt, M. Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J. Intern. Med. 2018, 284, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, S.; Wu, B.; Zhao, Y.; Liu, X.; Liang, Y.; Shao, X.; Holthöfer, H.; Zou, H. Association between metabolically unhealthy overweight/obesity and chronic kidney disease: The role of inflammation. Diabetes Metab. 2014, 40, 423–430. [Google Scholar] [CrossRef]
- Rashed, L.A.; Elattar, S.; Eltablawy, N.; Ashour, H.; Mahmoud, L.M.; El-Esawy, Y. Mesenchymal stem cells pretreated with melatonin ameliorate kidney functions in a rat model of diabetic nephropathy. Biochem. Cell Biol. 2018, 96, 564–571. [Google Scholar] [CrossRef]
- Rüster, C.; Wolf, G. Adipokines promote chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28 (Suppl. 4), iv8–iv14. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Carrero, J.J.; Stenvinkel, P. Inflammation as a risk factor and target for therapy in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, S.D.; Kirwan, J.P.; Remer, E.M.; Schneider, E.; Addeman, B.; Arrigain, S.; Horwitz, E.; Fink, J.C.; Lash, J.P.; McKenzie, C.A.; et al. Adiposity, physical function, and their associations with insulin resistance, inflammation, and adipokines in CKD. Am. J Kidney Dis. 2021, 77, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and progression of CKD: The CRIC study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin-Kelley, V.E.; Jevnikar, A.M. Antigen presentation by renal tubular epithelial cells. J. Am. Soc. Nephrol. 1991, 2, 13–26. [Google Scholar] [CrossRef]
- Declèves, A.-E.; Mathew, A.V.; Cunard, R.; Sharma, K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 2011, 22, 1846–1855. [Google Scholar] [CrossRef] [Green Version]
- Pruijm, M.; Ponte, B.; Vollenweider, P.; Mooser, V.; Paccaud, F.; Waeber, G.; Marques-Vidal, P.; Burnier, M.; Bochud, M. Not all inflammatory markers are linked to kidney function: Results from a population-based study. Am. J. Nephrol. 2012, 35, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, J.; Gai, Z.; Kullak-Ublick, G.A.; Liu, Z. TNF-α Deficiency prevents renal inflammation and oxidative stress in obese mice. Kidney Blood Press. Res. 2017, 42, 416–427. [Google Scholar] [CrossRef]
- Wen, Y.; Lu, X.; Ren, J.; Privratsky, J.R.; Yang, B.; Rudemiller, N.P.; Zhang, J.; Griffiths, R.; Jain, M.K.; Nedospasov, S.A.; et al. KLF4 in macrophages attenuates tnf-mediated kidney injury and fibrosis. J. Am. Soc. Nephrol. 2019, 30, 1925–1938. [Google Scholar] [CrossRef]
- Therrien, F.J.; Agharazii, M.; Lebel, M.; Larivière, R. Neutralization of tumor necrosis factor-alpha reduces renal fibrosis and hypertension in rats with renal failure. Am. J. Nephrol. 2012, 36, 151–161. [Google Scholar] [CrossRef]
- Eo, H.; Park, J.E.; Jeon, Y.-J.; Lim, Y. Ameliorative effect of ecklonia cava polyphenol extract on renal inflammation associated with aberrant energy metabolism and oxidative stress in high fat diet-induced obese mice. J. Agric. Food Chem. 2017, 65, 3811–3818. [Google Scholar] [CrossRef]
- Kizer, J.R. Adiponectin, cardiovascular disease, and mortality: Parsing the dual prognostic implications of a complex adipokine. Metabolism 2014, 63, 1079–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, T.; Mita, A.; Minami, K.; Hosooka, T.; Kitazawa, S.; Takahashi, K.; Tamori, Y.; Yokoi, N.; Watanabe, M.; Matsuo, E.-I.; et al. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 2012, 15, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.H.; Oh, T.R.; Choi, H.S.; Kim, C.S.; Ma, S.K.; Oh, K.H.; Ahn, C.; Kim, S.W.; Bae, E.H. High serum adiponectin as a biomarker of renal dysfunction: Results from the KNOW-CKD study. Sci. Rep. 2020, 10, 5598. [Google Scholar] [CrossRef]
- Kim-Mitsuyama, S.; Soejima, H.; Yasuda, O.; Node, K.; Jinnouchi, H.; Yamamoto, E.; Sekigami, T.; Ogawa, H.; Matsui, K. Total adiponectin is associated with incident cardiovascular and renal events in treated hypertensive patients: Subanalysis of the ATTEMPT-CVD randomized trial. Sci. Rep. 2019, 9, 16589. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Carrero, J.J.; Lindholm, B.; Stenvinkel, P. The complex role of adiponectin in chronic kidney disease. Biochimie 2012, 94, 2150–2156. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, C.W. Mechanisms of adiponectin action: Implication of adiponectin receptor agonism in diabetic kidney disease. Int. J. Mol. Sci. 2019, 20, 1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Russa, D.; Marrone, A.; Mandalà, M.; Macirella, R.; Pellegrino, D. Antioxidant/anti-inflammatory effects of caloric restriction in an aged and obese rat model: The role of adiponectin. Biomedicines 2020, 8, 532. [Google Scholar] [CrossRef]
- Mao, X.; Kikani, C.K.; Riojas, R.A.; Langlais, P.; Wang, L.; Ramos, F.J.; Fang, Q.; Christ-Roberts, C.Y.; Hong, J.Y.; Kim, R.-Y.; et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 2006, 8, 516–523. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Pessoa, E.D.A.; Convento, M.B.; Castino, B.; Leme, A.M.; de Oliveira, A.S.; Aragão, A.; Fernandes, S.M.; Carbonel, A.; Dezoti, C.; Vattimo, M.D.F.; et al. Beneficial effects of isoflavones in the kidney of obese rats are mediated by PPAR-gamma expression. Nutrients 2020, 12, 1624. [Google Scholar] [CrossRef]
- Declèves, A.-E.; Sharma, K. Obesity and kidney disease: Differential effects of obesity on adipose tissue and kidney inflammation and fibrosis. Curr. Opin. Nephrol. Hypertens. 2015, 24, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Roberts-Toler, C.; O’Neill, B.T.; Cypess, A.M. Diet-induced obesity causes insulin resistance in mouse brown adipose tissue. Obesity 2015, 23, 1765–1770. [Google Scholar] [CrossRef]
- Câmara, N.O.S.; Iseki, K.; Kramer, H.; Liu, Z.-H.; Sharma, K. Kidney disease and obesity: Epidemiology, mechanisms and treatment. Nat. Rev. Nephrol. 2017, 13, 181–190. [Google Scholar] [CrossRef]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Zelnick, L.R.; Robinson, N.R.; Hung, A.M.; Kestenbaum, B.; Utzschneider, K.M.; Kahn, S.E.; de Boer, I.H. Chronic kidney disease and obesity bias surrogate estimates of insulin sensitivity compared with the hyperinsulinemic euglycemic clamp. Am. J. Physiol. Metab. 2017, 312, E175–E182. [Google Scholar] [CrossRef]
- De Boer, I.H.; Zelnick, L.; Afkarian, M.; Ayers, E.; Curtin, L.; Himmelfarb, J.; Ikizler, T.A.; Kahn, S.E.; Kestenbaum, B.; Utzschneider, K. Impaired glucose and insulin homeostasis in moderate-severe CKD. J. Am. Soc. Nephrol. 2016, 27, 2861–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Wang, Q.; Yang, X. Fu brick tea alleviates chronic kidney disease of rats with high fat diet consumption through attenuating insulin resistance in skeletal muscle. J. Agric. Food Chem. 2019, 67, 2839–2847. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.-L.; Lin, Y.-J.; Sheen, J.-M.; Yu, H.-R.; Tiao, M.-M.; Chen, C.-C.; Tsai, C.-C.; Huang, L.-T.; Hsu, C.-N. High Fat diets sex-specifically affect the renal transcriptome and program obesity, kidney injury, and hypertension in the offspring. Nutrients 2017, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, N.; Ishigaki, S.; Isobe, S. The pivotal role of melatonin in ameliorating chronic kidney disease by suppression of the renin-angiotensin system in the kidney. Hypertens. Res. 2019, 42, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Passos-Silva, D.G.; Brandan, E.; Santos, R.A.S. Angiotensins as therapeutic targets beyond heart disease. Trends Pharmacol. Sci. 2015, 36, 310–320. [Google Scholar] [CrossRef]
- Cole, B.K.; Keller, S.R.; Wu, R.; Carter, J.D.; Nadler, J.L.; Nunemaker, C.S. Valsartan protects pancreatic islets and adipose tissue from the inflammatory and metabolic consequences of a high-fat diet in mice. Hypertension 2010, 55, 715–721. [Google Scholar] [CrossRef]
- Ma, L.-J.; Corsa, B.A.; Zhou, J.; Yang, H.; Li, H.; Tang, Y.-W.; Babaev, V.R.; Major, A.S.; Linton, M.F.; Fazio, S.; et al. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am. J. Physiol. Renal Physiol. 2011, 300, F1203–F1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.; Liu, J.; Niu, J.; Zhang, Y.; Shen, W.; Luo, C.; Liu, Y.; Li, C.; Li, H.; Yang, P.; et al. Wnt/β-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell. 2019, 18, e13004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelis, D.d.F.; Freitas, D.F.d.; Machado, A.S.; Crespo, T.S.; Santos, S.H.S. Angiotensin-(1-7), adipokines and inflammation. Metabolism 2019, 95, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hall, J.E.; Lu, D.; Lin, L.; Manning, R.D.; Cheng, L.; Gomez-Sanchez, C.E.; Juncos, L.A.; Liu, R. Aldosterone blunts tubuloglomerular feedback by activating macula densa mineralocorticoid receptors. Hypertension 2012, 59, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T. Mechanism of salt-sensitive hypertension: Focus on adrenal and sympathetic nervous systems. J. Am. Soc. Nephrol. 2014, 25, 1148–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-H.; Suendermann, C.; Steuer, A.E.; Pacheco Lopez, G.; Odermatt, A.; Faresse, N.; Henneberg, M.; Langhans, W. Aldosterone deficiency in mice burdens respiration and accentuates diet-induced hyperinsulinemia and obesity. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in kidney, heart, and skeletal muscle dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [Green Version]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D.; Stec, D.E. Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Renal Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M. Lipotoxicity. Kidney Int. 2006, 70, 1560–1566. [Google Scholar] [CrossRef] [Green Version]
- Katsoulieris, E.; Mabley, J.G.; Samai, M.; Sharpe, M.A.; Green, I.C.; Chatterjee, P.K. Lipotoxicity in renal proximal tubular cells: Relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic. Biol. Med. 2010, 48, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.R.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Kim, Y.; Choi, B.S.; Kim, Y.-S.; Kim, H.W.; Lim, K.-M.; Kim, M.J.; et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism 2018, 85, 348–360. [Google Scholar] [CrossRef]
- Zhu, Q.; Scherer, P.E. Immunologic and endocrine functions of adipose tissue: Implications for kidney disease. Nat. Rev. Nephrol. 2018, 14, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Varghese, Z.; Ruan, X.Z. The molecular pathogenic role of inflammatory stress in dysregulation of lipid homeostasis and hepatic steatosis. Genes Dis. 2014, 1, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Yokoi, H.; Yanagita, M. Targeting the fatty acid transport protein CD36, a class B scavenger receptor, in the treatment of renal disease. Kidney Int. 2016, 89, 740–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Xiao, Y.; Luo, X.; Zhao, Y.; Zhao, L.; Wang, Y.; Wu, T.; Wei, L.; Chen, Y. Inflammatory stress promotes the development of obesity-related chronic kidney disease via CD36 in mice. J. Lipid Res. 2017, 58, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, E.; Gutiérrez, E.; Morales, E.; Hernández, E.; Andres, A.; Bello, I.; Díaz-González, R.; Leiva, O.; Praga, M. Factors influencing the progression of renal damage in patients with unilateral renal agenesis and remnant kidney. Kidney Int. 2005, 68, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallamaci, F.; Ruggenenti, P.; Perna, A.; Leonardis, D.; Tripepi, R.; Tripepi, G.; Remuzzi, G.; Zoccali, C. ACE inhibition is renoprotective among obese patients with proteinuria. J. Am. Soc. Nephrol. 2011, 22, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, M.; Liu, P.; Wang, Y.; Zhang, H.; Li, H.; Yang, S.; Song, Y.; Yin, Y.; Gao, L.; et al. Telmisartan ameliorates nephropathy in metabolic syndrome by reducing leptin release from perirenal adipose tissue. Hypertension 2016, 68, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomback, A.S.; Muskala, P.; Bald, E.; Chwatko, G.; Nowicki, M. Low-dose spironolactone, added to long-term ACE inhibitor therapy, reduces blood pressure and urinary albumin excretion in obese patients with hypertensive target organ damage. Clin. Nephrol. 2009, 72, 449–456. [Google Scholar] [CrossRef]
- Strojek, K.; Yoon, K.H.; Hruba, V.; Elze, M.; Langkilde, A.M.; Parikh, S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: A randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2011, 13, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.D.; Skene, D.J. 60 years of neuroendocrinology: Regulation of mammalian neuroendocrine physiology and rhythms by melatonin. J. Endocrinol. 2015, 226, T187–T198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigaki, S.; Ohashi, N.; Matsuyama, T.; Isobe, S.; Tsuji, N.; Iwakura, T.; Fujikura, T.; Tsuji, T.; Kato, A.; Miyajima, H.; et al. Melatonin ameliorates intrarenal renin-angiotensin system in a 5/6 nephrectomy rat model. Clin. Exp. Nephrol. 2018, 22, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozbek, E.; Ilbey, Y.O.; Ozbek, M.; Simsek, A.; Cekmen, M.; Somay, A. Melatonin attenuates unilateral ureteral obstruction-induced renal injury by reducing oxidative stress, iNOS, MAPK, and NF-kB expression. J. Endourol. 2009, 23, 1165–1173. [Google Scholar] [CrossRef]
- Hussein, M.R.; Ahmed, O.G.; Hassan, A.F.; Ahmed, M.A. Intake of melatonin is associated with amelioration of physiological changes, both metabolic and morphological pathologies associated with obesity: An animal model. Int. J. Exp. Pathol. 2007, 88, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Erşahin, M.; Sehirli, O.; Toklu, H.Z.; Süleymanoglu, S.; Emekli-Alturfan, E.; Yarat, A.; Tatlidede, E.; Yeğen, B.C.; Sener, G. Melatonin improves cardiovascular function and ameliorates renal, cardiac and cerebral damage in rats with renovascular hypertension. J. Pineal Res. 2009, 47, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, K.; Focht, D.; Sierakowski, B.; Lewandowski, K.; Orlowska, M.; Usarek, M. NADPH oxidase inhibitor, apocynin, improves renal glutathione status in Zucker diabetic fatty rats: A comparison with melatonin. Chem. Biol. Interact. 2014, 218, 12–19. [Google Scholar] [CrossRef]
- La Russa, D.; Giordano, F.; Marrone, A.; Parafati, M.; Janda, E.; Pellegrino, D. Oxidative Imbalance and kidney damage in cafeteria diet-induced rat model of metabolic syndrome: Effect of bergamot polyphenolic fraction. Antioxidants 2019, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikorska, D.; Grzymislawska, M.; Roszak, M.; Gulbicka, P.; Korybalska, K.; Witowski, J. Simple obesity and renal function. J. Physiol. Pharmacol. 2017, 68, 175–180. [Google Scholar]
- Tsuboi, N.; Okabayashi, Y.; Shimizu, A.; Yokoo, T. The Renal Pathology of Obesity. Kidney Int. Rep. 2017, 2, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Remuzzi, G.; Ruggenenti, P.; Perico, N. Chronic renal diseases: Renoprotective benefits of renin-angiotensin system inhibition. Ann. Intern. Med. 2002, 136, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Adler, G.K. Aldosterone and the mineralocorticoid receptor: Risk factors for cardiometabolic disorders. Curr. Hypertens. Rep. 2015, 17, 52. [Google Scholar] [CrossRef]
- Praga, M.; Morales, E. The fatty kidney: Obesity and renal disease. Nephron 2017, 136, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Jaikumkao, K.; Pongchaidecha, A.; Chatsudthipong, V.; Chattipakorn, S.C.; Chattipakorn, N.; Lungkaphin, A. The roles of sodium-glucose cotransporter 2 inhibitors in preventing kidney injury in diabetes. Biomed. Pharmacother. 2017, 94, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Tsogka, K.; Nikolaou, E.; Liakopoulos, V.; Stefanidis, I. A unifying model of glucotoxicity in human renal proximal tubular epithelial cells and the effect of the SGLT2 inhibitor dapagliflozin. Int. Urol. Nephrol. 2020, 52, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Fioretto, P.; Tang, W.; List, J.F. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014, 85, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 protein expression is increased in human diabetic nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z.I. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: Cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Zingerman, B.; Herman-Edelstein, M.; Erman, A.; Bar Sheshet Itach, S.; Ori, Y.; Rozen-Zvi, B.; Gafter, U.; Chagnac, A. Effect of acetazolamide on obesity-induced glomerular hyperfiltration: A randomized controlled trial. PLoS ONE 2015, 10, e0137163. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Chen, S.; Zeng, S.; Zhao, Y.; Zhu, C.; Deng, B.; Zhu, G.; Yin, Y.; Wang, W.; Hardeland, R.; et al. Melatonin in macrophage biology: Current understanding and future perspectives. J. Pineal Res. 2019, 66, e12547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekmekcioglu, C. Melatonin receptors in humans: Biological role and clinical relevance. Biomed. Pharmacother. 2006, 60, 97–108. [Google Scholar] [CrossRef]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, functions and therapeutic benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Prado, N.J.; Ferder, L.; Manucha, W.; Diez, E.R. Anti-inflammatory effects of melatonin in obesity and hypertension. Curr. Hypertens. Rep. 2018, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Cipolla-Neto, J.; Amaral, F.G.; Afeche, S.C.; Tan, D.X.; Reiter, R.J. Melatonin, energy metabolism, and obesity: A review. J. Pineal Res. 2014, 56, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Buonfiglio, D.; Parthimos, R.; Dantas, R.; Cerqueira Silva, R.; Gomes, G.; Andrade-Silva, J.; Ramos-Lobo, A.; Amaral, F.G.; Matos, R.; Sinésio, J.; et al. Melatonin absence leads to long-term leptin resistance and overweight in rats. Front. Endocrinol. 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; Cano, P.; Jiménez-Ortega, V.; Esquifino, A.I. Melatonin and the metabolic syndrome: Physiopathologic and therapeutical implications. Neuroendocrinology 2011, 93, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Laermans, J.; Depoortere, I. Chronobesity: Role of the circadian system in the obesity epidemic. Obes. Rev. 2016, 17, 108–125. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J. Pineal Res. 2017, 63, e12414. [Google Scholar] [CrossRef]
- Ke, B.; Shen, W.; Fang, X.; Wu, Q. The NLPR3 inflammasome and obesity-related kidney disease. J. Cell. Mol. Med. 2018, 22, 16–24. [Google Scholar] [CrossRef]
- Wang, J.; Wen, Y.; Lv, L.-L.; Liu, H.; Tang, R.-N.; Ma, K.-L.; Liu, B.-C. Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol. Sin. 2015, 36, 821–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.-Y.; Tang, L.-Q. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol. Res. 2016, 114, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Chintam, K.; Chang, A.R. Strategies to treat obesity in patients with CKD. Am. J. Kidney Dis. 2021, 77, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. Mechanisms and treatment of CKD. J. Am. Soc. Nephrol. 2012, 23, 1917–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, M.S.; Lewis, J.; Huntsberry, A.M.; Dea, L.; Portillo, I. Efficacy and renal outcomes of SGLT2 inhibitors in patients with type 2 diabetes and chronic kidney disease. Postgrad. Med. 2019, 131, 31–42. [Google Scholar] [CrossRef]
- Onaolapo, A.Y.; Adebisi, E.O.; Adeleye, A.E.; Olofinnade, A.T.; Onaolapo, O.J. Dietary Melatonin Protects against Behavioural, Metabolic, Oxidative, and Organ Morphological Changes in Mice that are Fed High-Fat, High-Sugar Diet. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 570–583. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Sun, N.; Liu, X.; Song, E.; Zhang, Z.; Wen, J.; Zheng, T. Melatonin therapy protects against renal injury before and after release of bilateral ureteral obstruction in rats. Life Sci. 2019, 229, 104–115. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Kidney Injury | Pathological Damage in Kidney | References |
|---|---|---|
| Glomerular injury | Global or focal segmental glomerulosclerosis | [20,21] |
| Glomerulomegaly (glomerular volume > 3.27 × 106 μm3) with or without FSGS | [22,23,24] | |
| Low glomerular density | [23] | |
| Glomerulosclerosis | [21] | |
| Glomerular filtration barrier defect | [25] | |
| Renal tubular injury | Tubular atrophy | [19] |
| Proximal tubular epithelial hypertrophy | [26] | |
| Tubular cell apoptosis ↑ | [25] | |
| Oxidative stress in tubular cells | ||
| Others | The measured parenchymal volume and the estimated cortical volume of the kidneys ↑ | [21] |
| ·Oxidative stress | ||
| ·Enhanced renal lipid deposition | ||
| ·Renal TGF-β1 and collagen type IV expression ↑ | ||
| Glomerular and tubular lesion scores ↑ |
| Animal/Model | Drug/Dose | Renal Function | Kidney Injury | Reference |
|---|---|---|---|---|
| Human Obesity FSG | ACEI | Proteinuria ↓ | Glomerulomegaly | [20] |
| Human Obesity RK or URA | ACEI | Proteinuria/renal insufficiency, serum creatinine ↓ | N/A | [100] |
| Human Obesity ESRD | ACEI | Proteinuria ↓ BP burden → | N/A | [101] |
| Male Wistar rats Obesity | ACEI, telmisartan, (8 mg/kg/d, gastriclavage) | N/A | Local expression of adipogenesis markers and adiponectin ↑ IL-6, MCP-1 ↓ | [102] |
| Human Obesity | Low-dose spironolactone + ACEI | Blood pressures, proteinuria ↓ | N/A | [103] |
| Human Type 2 diabetes CKD | SGLT2 inhibitors, dapagliflozin (5 and 10 mg) | Blood pressures, proteinuria ↓ Serum creatinine ↑ | N/A | [104] |
| Akita mice Diabetic | SGLT2 inhibitors, empagliflozin (300 mg/kg) | Systolic blood pressure ↓ | GLUT1/GLUT2, inflammation, CD14,IL-6,TIMP2 ↓ | [105] |
| Sprague-Dawleyrats 5/6 Nx CKD | Melatonin, (10 mg/100 mL, drink) | Blood pressure ↓ | RAS, oxidative stress, Interstitial fibrosis ↓ Antioxidant activity ↑ | [106] |
| Wistar Albino rat Unilateral ureteral obstruction (UUO) | Melatonin (1 mg/kg/day, intraperitoneal) | N/A | Oxidative stress, iNOS, p38-MAPK, NF-kB ↓ The development of leukocyte infiltration and interstitial fibrosis ↓ | [107] |
| Boscat white rabbits Obesity | Melatonin, 1 mg/kg (sub-cutaneously) | Blood pressure, serum lipids, blood glucose, atherogenic index ↓ | GSH-PX ↑ | [108] |
| Wistar albino rats Renovascular hypertension | Melatonin (10 mg/kg/day) | Blood pressure ↓ | Oxidative injury, plasma LDH, CK and ADMA levels ↓ | [109] |
| Zücker rats Obesity Diabetic | Melatonin, (2 g/L and 20 mg/L, drink) | Proteinuria ↓ | NOx, HFR, GSH/GSSG ↓ | [110] |
| Wistar albino rats Diabetic nephropathy | MSCs + Melatonin (5 µM, in vitro) | N/A | Anti-inflammatory Anti-oxidation TNF-α, TGF-β1 ↓ IL-10, SOD, Beclin-1, glomerular sclerosis ↑ | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Wang, Z.; Chen, Y.; Dong, Y. Kidney Damage Caused by Obesity and Its Feasible Treatment Drugs. Int. J. Mol. Sci. 2022, 23, 747. https://doi.org/10.3390/ijms23020747
Wang M, Wang Z, Chen Y, Dong Y. Kidney Damage Caused by Obesity and Its Feasible Treatment Drugs. International Journal of Molecular Sciences. 2022; 23(2):747. https://doi.org/10.3390/ijms23020747
Chicago/Turabian StyleWang, Meihui, Zixu Wang, Yaoxing Chen, and Yulan Dong. 2022. "Kidney Damage Caused by Obesity and Its Feasible Treatment Drugs" International Journal of Molecular Sciences 23, no. 2: 747. https://doi.org/10.3390/ijms23020747
APA StyleWang, M., Wang, Z., Chen, Y., & Dong, Y. (2022). Kidney Damage Caused by Obesity and Its Feasible Treatment Drugs. International Journal of Molecular Sciences, 23(2), 747. https://doi.org/10.3390/ijms23020747