
The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets


 , ,
, , 
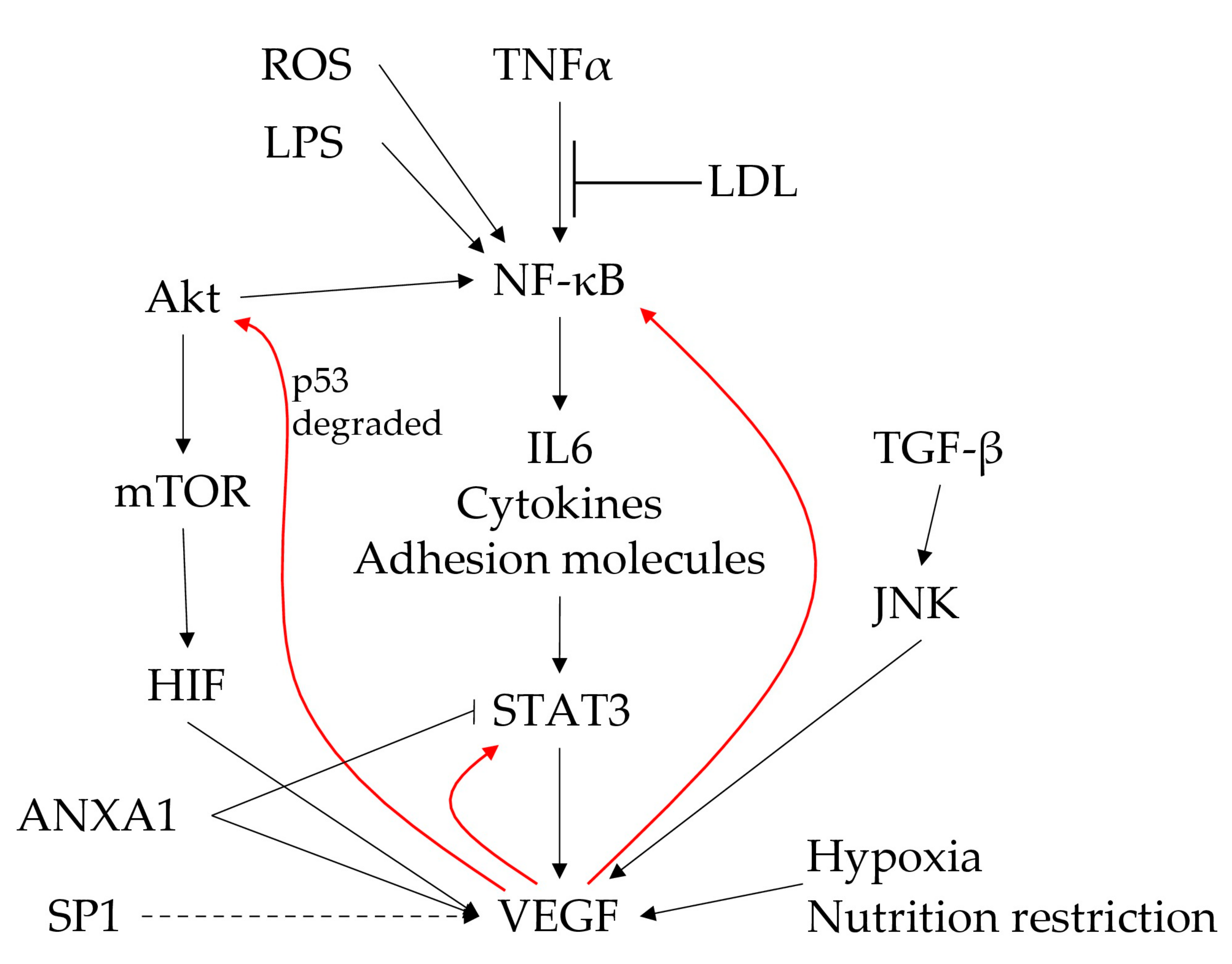{kind=link}
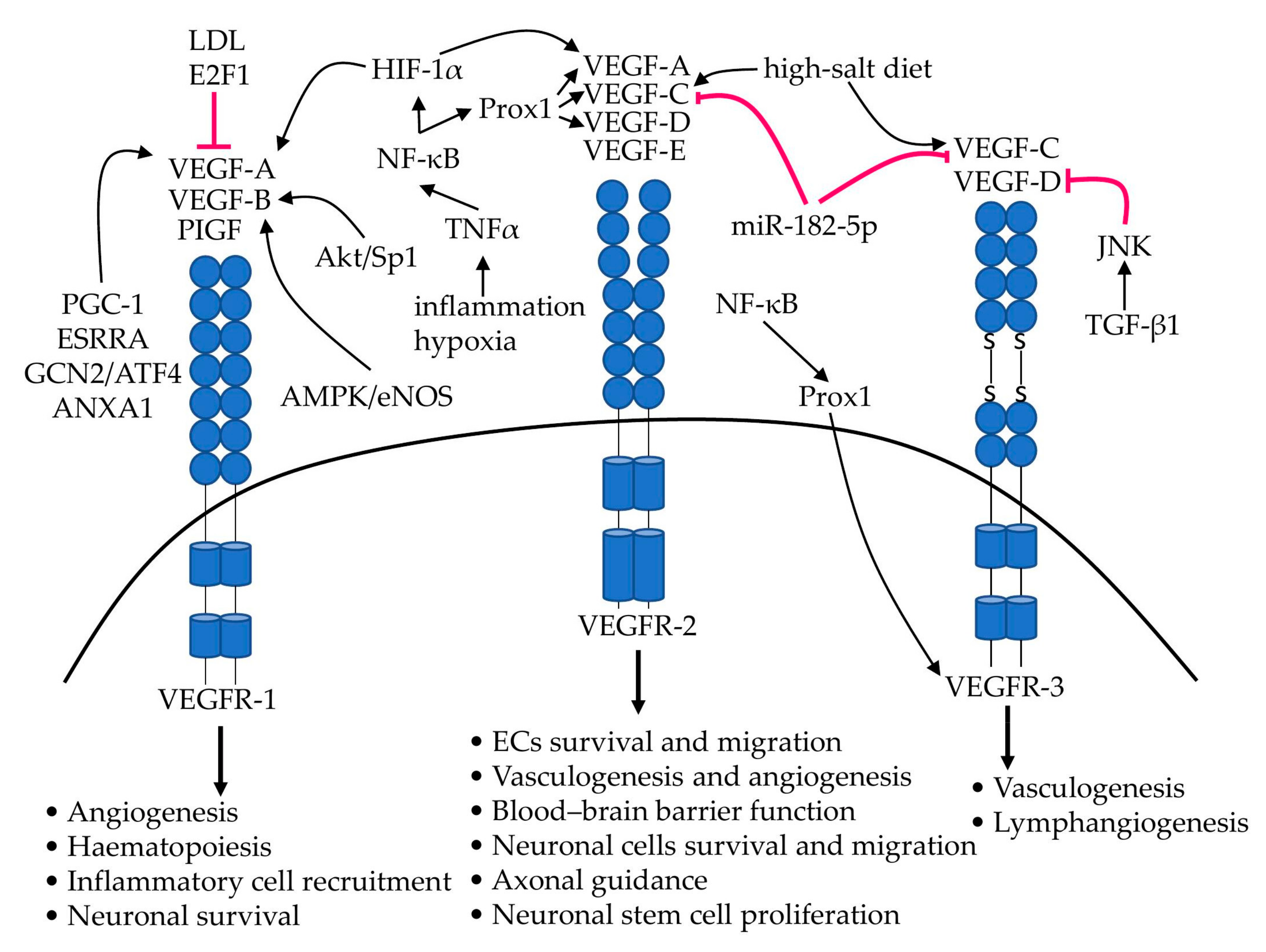{kind=link}
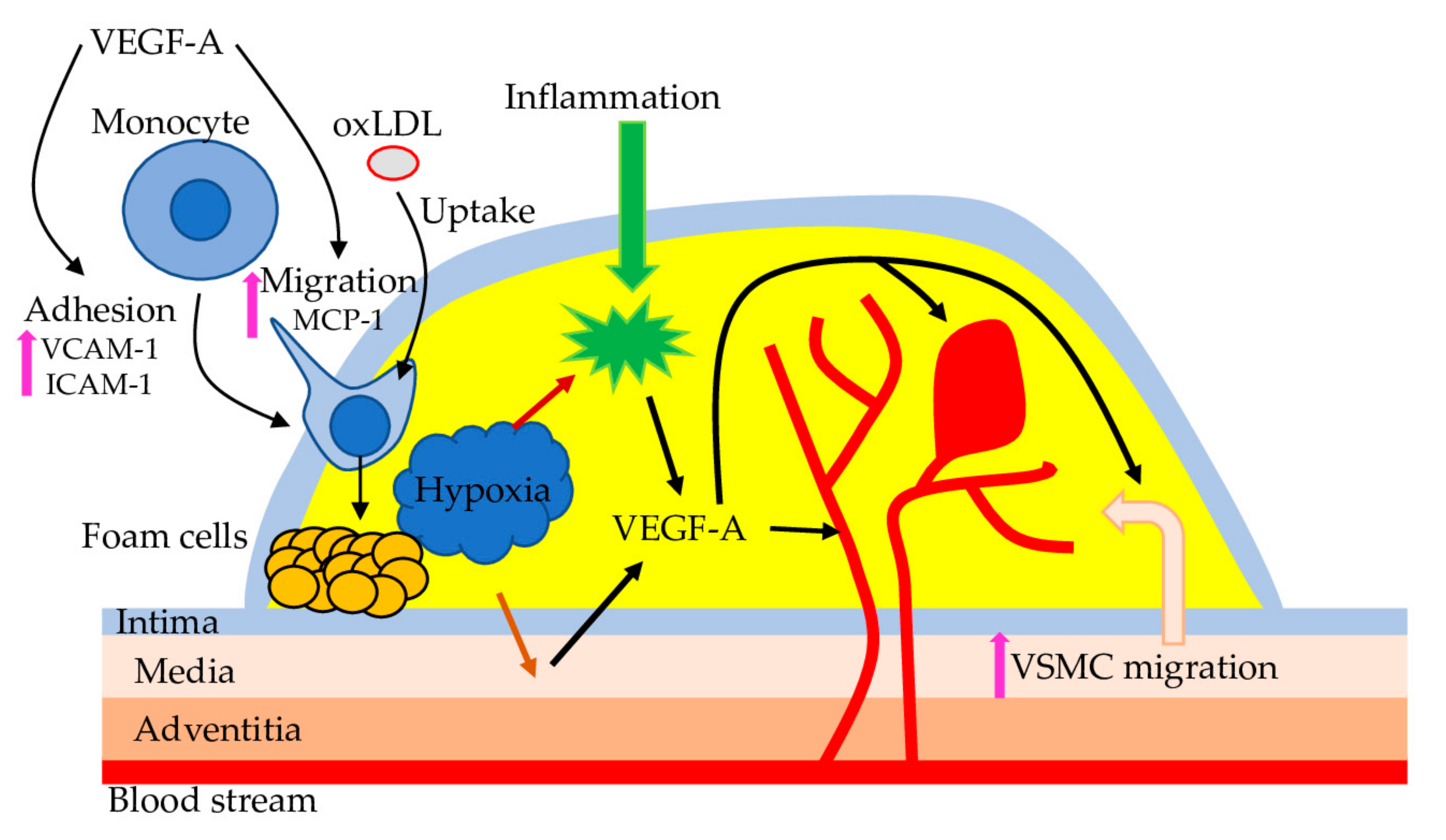{kind=link}
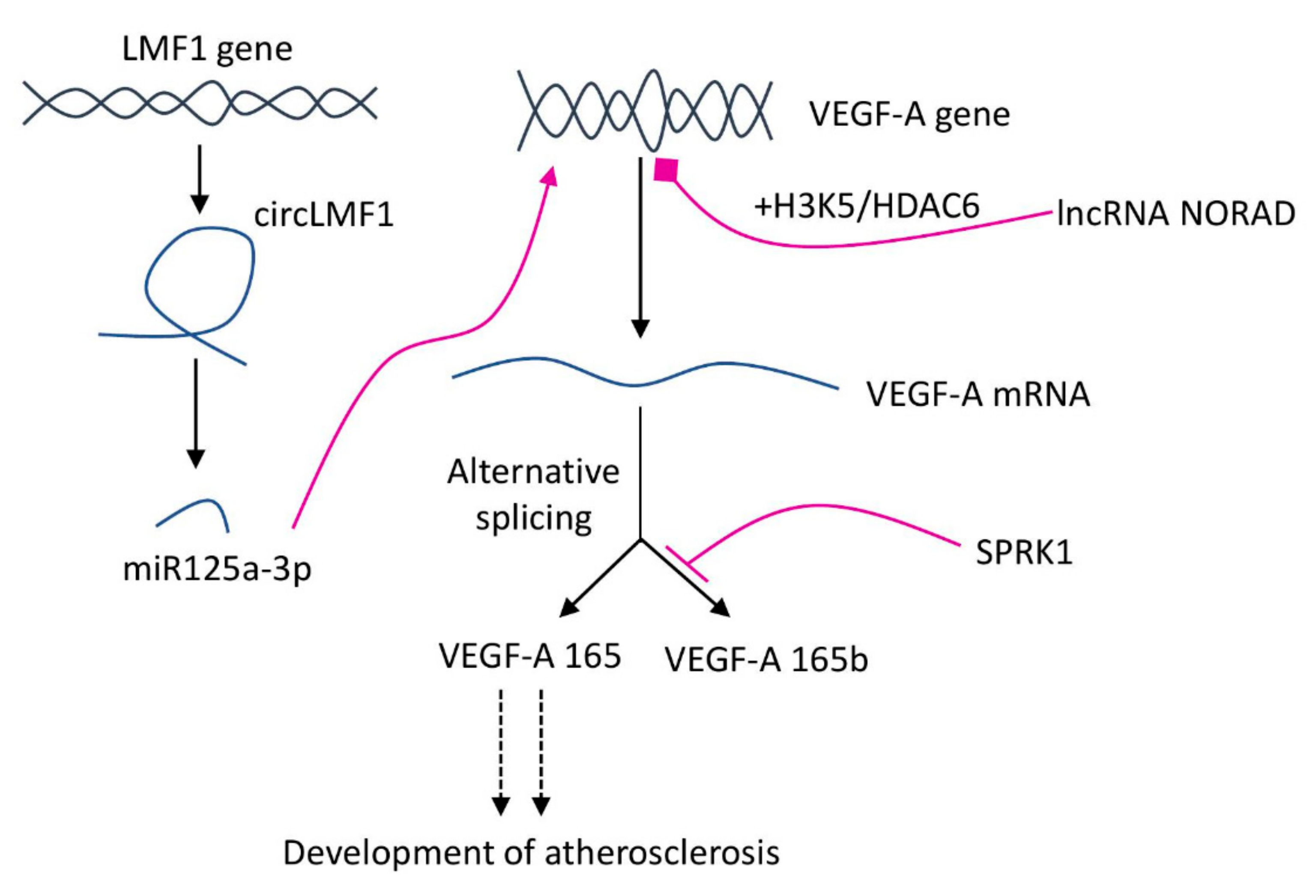{kind=link}
Abstract
:1. Introduction
2. VEGFs Regulation and Functions in CVDs
2.1. VEGF-A
2.2. VEGF-B
2.3. VEGF-C
2.4. VEGF-D
3. The Role of VEGFs in Lipid Metabolism
3.1. VEGF-A
3.2. VEGF-B
3.3. VEGF-C
3.4. VEGF-D
4. Atherosclerosis
5. VEGF as a Therapeutic Target
5.1. Experimental Procedures
5.2. Natural Compounds
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin-converting enzyme 2 |
| ADSC | adipose-derived stem cell |
| AECs | aortic endothelial cells |
| AMPK | 5′AMP-Activated Protein Kinase Catalytic Subunit Alpha-2/l |
| CAD | coronary artery disease |
| CM | chylomicrons |
| CRP | C-reactive protein |
| CVDs | cardiovascular diseases |
| ECs | endothelial cells |
| eNOS | Nitric Oxide Synthase, Endothelia |
| HIF-1α | Hypoxia Inducible Factor 1 Subunit Alpha |
| LDL | low-density lipoprotein |
| LECs | lymphatic endothelial cells |
| LMF1 | lipase maturation factor 1 |
| LPL | plasma lipoprotein lipase |
| NO | nitric oxide |
| PAD | peripheral arterial disease |
| PF | Paeoniflorin |
| SDF1 | Stromal Cell-Derived Factor 1 |
| STAT3 | Signal Transducer And Activator Of Transcription 3 |
| TC | total cholesterol |
| TG | triglyceride |
| TGF-β1 | transforming growth factor-β1 |
| TMP | Tetramethylpyrazine |
| VCAM-1 | Vascular Cell Adhesion Molecule 1 |
| VECs | vascular endothelial cell |
| VEGF | vascular endothelial growth factor |
| VEGFR1 | Vascular Endothelial Growth Factor Receptor |
| VSP | VEGF signalling pathway |
References
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor Cells Secrete a Vascular Permeability Factor That Promotes Accumulation of Ascites Fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Corlan, A.S.; Cîmpean, A.M.; Jitariu, A.-A.; Melnic, E.; Raica, M. Endocrine Gland-Derived Vascular Endothelial Growth Factor/Prokineticin-1 in Cancer Development and Tumor Angiogenesis. Int. J. Endocrinol. 2017, 2017, 3232905. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular Endothelial Growth Factor (VEGF)—Key Factor in Normal and Pathological Angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar] [PubMed]
- Hokkanen, K.; Tirronen, A.; Ylä-Herttuala, S. Intestinal Lymphatic Vessels and Their Role in Chylomicron Absorption and Lipid Homeostasis. Curr. Opin. Lipidol. 2019, 30, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Milasan, A.; Smaani, A.; Martel, C. Early Rescue of Lymphatic Function Limits Atherosclerosis Progression in Ldlr Mice. Atherosclerosis 2019, 283, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [Green Version]
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. VEGF Signals through ATF6 and PERK to Promote Endothelial Cell Survival and Angiogenesis in the Absence of ER Stress. Mol. Cell 2014, 54, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-Angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- Mukwaya, A.; Jensen, L.; Lagali, N. Relapse of Pathological Angiogenesis: Functional Role of the Basement Membrane and Potential Treatment Strategies. Exp. Mol. Med. 2021, 53, 189–201. [Google Scholar] [CrossRef]
- Ganta, V.C.; Choi, M.; Kutateladze, A.; Annex, B.H. VEGF165b Modulates Endothelial VEGFR1–STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease. Circ. Res. 2017, 120, 282–295. [Google Scholar] [CrossRef] [Green Version]
- Braile, M.; Marcella, S.; Cristinziano, L.; Galdiero, M.R.; Modestino, L.; Ferrara, A.L.; Varricchi, G.; Marone, G.; Loffredo, S. VEGF-A in Cardiomyocytes and Heart Diseases. Int. J. Mol. Sci. 2020, 21, 5294. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Zheng, X.; Yang, Y.; Yao, G.; Ye, L.; Doeppner, T.R.; Hermann, D.M.; Wang, H.; Dai, Y. Impairment of Hypoxia-Induced Angiogenesis by LDL Involves a HIF-Centered Signaling Network Linking Inflammatory TNFα and Angiogenic VEGF. Aging 2019, 11, 328–349. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Foo, S.-Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-Independent Regulation of VEGF and Angiogenesis by the Transcriptional Coactivator PGC-1α. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longchamp, A.; Mirabella, T.; Arduini, A.; MacArthur, M.R.; Das, A.; Treviño-Villarreal, J.H.; Hine, C.; Ben-Sahra, I.; Knudsen, N.H.; Brace, L.E.; et al. Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 2018, 173, 117–129.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, B.; Leoni, G.; Hinkel, R.; Ormanns, S.; Paulin, N.; Ortega-Gomez, A.; Viola, J.R.; de Jong, R.; Bongiovanni, D.; Bozoglu, T.; et al. Pro-Angiogenic Macrophage Phenotype to Promote Myocardial Repair. J. Am. Coll. Cardiol. 2019, 73, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhou, J.; Cheng, M.; Boriboun, C.; Biyashev, D.; Wang, H.; Mackie, A.; Thorne, T.; Chou, J.; Wu, Y.; et al. E2F1 Suppresses Cardiac Neovascularization by Down-Regulating VEGF and PlGF Expression. Cardiovasc. Res. 2014, 104, 412–422. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Zhang, B.; Jiang, J.; Wang, Y.; Wu, Y. Up-Regulation of ANXA1 Suppresses Polymorphonuclear Neutrophil Infiltration and Myeloperoxidase Activity by Activating STAT3 Signaling Pathway in Rat Models of Myocardial Ischemia-Reperfusion Injury. Cell. Signal. 2019, 62, 109325. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Ojha, U.; Lee, Y.M. Pathological Angiogenesis and Inflammation in Tissues. Arch. Pharmacal Res. 2021, 44, 1–15. [Google Scholar] [CrossRef]
- Lv, Y.; Zhong, S.; Tang, H.; Luo, B.; Chen, S.-J.; Chen, L.; Zheng, F.; Zhang, L.; Wang, L.; Li, X.; et al. VEGF-A and VEGF-B Coordinate the Arteriogenesis to Repair the Infarcted Heart with Vagus Nerve Stimulation. Cell. Physiol. Biochem. 2018, 48, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Lal, N.; Puri, K.; Rodrigues, B. Vascular Endothelial Growth Factor B and Its Signaling. Front. Cardiovasc. Med. 2018, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, N.; Chiu, A.P.-L.; Wang, F.; Zhang, D.; Jia, J.; Wan, A.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Loss of VEGFB and Its Signaling in the Diabetic Heart Is Associated with Increased Cell Death Signaling. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1163–H1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Ren, J.; Li, Y.; Yang, G.; Kang, L.; Zhang, S.; Ma, C.; Li, J.; Liu, J.; Yang, L.; et al. Resveratrol Protects against Isoproterenol Induced Myocardial Infarction in Rats through VEGF-B/AMPK/ENOS/NO Signalling Pathway. Free. Radic. Res. 2019, 53, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Suzuki, M.; Matsuda, M.; Ajiro, Y.; Shinozaki, T.; Sakagami, S.; Yonezawa, K.; Shimizu, M.; Funada, J.; Takenaka, T.; et al. VEGF-C and Mortality in Patients With Suspected or Known Coronary Artery Disease. J. Am. Hear. Assoc. 2018, 7, e010355. [Google Scholar] [CrossRef] [Green Version]
- Houssari, M.; Dumesnil, A.; Tardif, V.; Kivelä, R.; Pizzinat, N.; Boukhalfa, I.; Godefroy, D.; Schapman, D.; Hemanthakumar, K.A.; Bizou, M.; et al. Lymphatic and Immune Cell Cross-Talk Regulates Cardiac Recovery After Experimental Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2020, 40, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.H.; Choe, K.; Hong, S.P.; Jeong, S.; Mäkinen, T.; Kim, K.S.; Alitalo, K.; Surh, C.D.; Koh, G.Y.; Song, J. Gut Microbiota Regulates Lacteal Integrity by Inducing VEGF-C in Intestinal Villus Macrophages. EMBO Rep. 2019, 20, e46927. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-H.; Zhou, X.; Ji, W.-J.; Liu, J.-X.; Sun, J.; Dong, Y.; Jiang, T.-M.; Li, Y.-M. VEGF-C-Mediated Cardiac Lymphangiogenesis in High Salt Intake Accelerated Progression of Left Ventricular Remodeling in Spontaneously Hypertensive Rats. Clin. Exp. Hypertens. 2017, 39, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wang, H.; Chen, X.; Liang, C.; Shang, W.; Wang, L.; Li, J.; Xu, D. MiR-182-5p Inhibits Colon Cancer Tumorigenesis, Angiogenesis, and Lymphangiogenesis by Directly Downregulating VEGF-C. Cancer Lett. 2020, 488, 18–26. [Google Scholar] [CrossRef]
- Muhammad, A.; Funmilola, A.; Aimola, I.A.; Ndams, I.S.; Inuwa, M.H.; Nok, A.J. Kolaviron Shows Anti-Proliferative Effect and down Regulation of Vascular Endothelial Growth Factor-C and Toll like Receptor-2 in Wuchereria Bancrofti Infected Blood Lymphocytes. J. Infect. Public Health 2017, 10, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Chen, Y.; Li, X.; Zhang, G.; Jin, D.; Zhao, H.; Su, Z.; Fan, Y. Norcantharidin Inhibits Lymphangiogenesis by Downregulating the Expression of VEGF-C and VEGF-D in Human Dermal Lymphatic Endothelial Cells in Vitro. Pharmacology 2015, 95, 362418. [Google Scholar] [CrossRef]
- Berntsson, J.; Smith, J.G.; Johnson, L.S.B.; Söderholm, M.; Borné, Y.; Melander, O.; Orho-Melander, M.; Nilsson, J.; Engström, G. Increased Vascular Endothelial Growth Factor D Is Associated with Atrial Fibrillation and Ischaemic Stroke. Heart 2019, 105, 553–558. [Google Scholar] [CrossRef]
- Wada, H.; Suzuki, M.; Matsuda, M.; Ajiro, Y.; Shinozaki, T.; Sakagami, S.; Yonezawa, K.; Shimizu, M.; Funada, J.; Takenaka, T.; et al. Distinct Characteristics of VEGF-D and VEGF-C to Predict Mortality in Patients With Suspected or Known Coronary Artery Disease. J. Am. Hear. Assoc. 2020, 9, e015761. [Google Scholar] [CrossRef] [PubMed]
- Morfoisse, F.; Renaud, E.; Hantelys, F.; Prats, A.-C.; Garmy-Susini, B. Role of Hypoxia and Vascular Endothelial Growth Factors in Lymphangiogenesis. Mol. Cell. Oncol. 2015, 2, e1024821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Jing, X.; Song, G.; Jie, L.; Shen, F. Prox1 Induces New Lymphatic Vessel Formation and Promotes Nerve Reconstruction in a Mouse Model of Sciatic Nerve Crush Injury. J. Anat. 2020, 237, 933–940. [Google Scholar] [CrossRef]
- Cui, Y.; Osorio, J.C.; Risquez, C.; Wang, H.; Shi, Y.; Gochuico, B.R.; Morse, D.; Rosas, I.O.; El-Chemaly, S. Transforming Growth Factor-Β1 Downregulates Vascular Endothelial Growth Factor-D Expression in Human Lung Fibroblasts via the Jun NH2-Terminal Kinase Signaling Pathway. Mol. Med. 2014, 20, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.; Achen, M. Emerging Roles for VEGF-D in Human Disease. Biomolecules 2018, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.E.; Kivelä, A.M.; Huusko, J.; Dijkstra, M.H.; Gurzeler, E.; Mäkinen, P.I.; Leppänen, P.; Olkkonen, V.M.; Eriksson, U.; Jauhiainen, M.; et al. The Effects of VEGF-A on Atherosclerosis, Lipoprotein Profile, and Lipoprotein Lipase in Hyperlipidaemic Mouse Models. Cardiovasc. Res. 2013, 99, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Schüler, R.; Seebeck, N.; Osterhoff, M.A.; Witte, V.; Flöel, A.; Busjahn, A.; Jais, A.; Brüning, J.C.; Frahnow, T.; Kabisch, S.; et al. VEGF and GLUT1 Are Highly Heritable, Inversely Correlated and Affected by Dietary Fat Intake: Consequences for Cognitive Function in Humans. Mol. Metab. 2018, 11, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular Endothelial Growth Factor B Controls Endothelial Fatty Acid Uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Moessinger, C.; Nilsson, I.; Muhl, L.; Zeitelhofer, M.; Heller Sahlgren, B.; Skogsberg, J.; Eriksson, U. VEGF-B Signaling Impairs Endothelial Glucose Transcytosis by Decreasing Membrane Cholesterol Content. EMBO Rep. 2020, 21, e49343. [Google Scholar] [CrossRef]
- Ning, F.C.; Jensen, N.; Mi, J.; Lindström, W.; Balan, M.; Muhl, L.; Eriksson, U.; Nilsson, I.; Nyqvist, D. VEGF-B Ablation in Pancreatic β-Cells Upregulates Insulin Expression without Affecting Glucose Homeostasis or Islet Lipid Uptake. Sci. Rep. 2020, 10, 923. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Shan, Z.; Wang, F.; Gao, X.; Tong, Y. Vascular Endothelial Growth Factor B Exerts Lipid-Lowering Effect by Activating AMPK via VEGFR1. Life Sci. 2021, 276, 119401. [Google Scholar] [CrossRef]
- Shew, T.; Wolins, N.E.; Cifarelli, V. VEGFR-3 Signaling Regulates Triglyceride Retention and Absorption in the Intestine. Front. Physiol. 2018, 9, 1783. [Google Scholar] [CrossRef] [PubMed]
- Karaman, S.; Hollmén, M.; Robciuc, M.R.; Alitalo, A.; Nurmi, H.; Morf, B.; Buschle, D.; Alkan, H.F.; Ochsenbein, A.M.; Alitalo, K.; et al. Blockade of VEGF-C and VEGF-D Modulates Adipose Tissue Inflammation and Improves Metabolic Parameters under High-Fat Diet. Mol. Metab. 2015, 4, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, H.; Saharinen, P.; Zarkada, G.; Zheng, W.; Robciuc, M.R.; Alitalo, K. VEGF-C Is Required for Intestinal Lymphatic Vessel Maintenance and Lipid Absorption. EMBO Mol. Med. 2015, 7, 1418–1425. [Google Scholar] [CrossRef]
- Karaman, S.; Hollmén, M.; Yoon, S.-Y.; Alkan, H.F.; Alitalo, K.; Wolfrum, C.; Detmar, M. Transgenic Overexpression of VEGF-C Induces Weight Gain and Insulin Resistance in Mice. Sci. Rep. 2016, 6, 31566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirronen, A.; Vuorio, T.; Kettunen, S.; Hokkanen, K.; Ramms, B.; Niskanen, H.; Laakso, H.; Kaikkonen, M.U.; Jauhiainen, M.; Gordts, P.L.S.M.; et al. Deletion of Lymphangiogenic and Angiogenic Growth Factor VEGF-D Leads to Severe Hyperlipidemia and Delayed Clearance of Chylomicron Remnants. Arter. Thromb. Vasc. Biol. 2018, 38, 2327–2337. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Barajas, S.; Lammoglia, G.M.; Reyna, A.J.; Morley, T.S.; Johnson, J.A.; Scherer, P.E.; Rutkowski, J.M. Vascular Endothelial Growth Factor–D (VEGF-D) Overexpression and Lymphatic Expansion in Murine Adipose Tissue Improves Metabolism in Obesity. Am. J. Pathol. 2019, 189, 924–939. [Google Scholar] [CrossRef] [Green Version]
- Kobiyama, K.; Ley, K. Atherosclerosis: A Chronic Inflammatory Disease With an Autoimmune Component. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Role of Lipids and Intraplaque Hypoxia in the Formation of Neovascularization in Atherosclerosis. Ann. Med. 2017, 49, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Milutinović, A.; Šuput, D.; Zorc-Pleskovič, R. Pathogenesis of Atherosclerosis in the Tunica Intima, Media, and Adventitia of Coronary Arteries: An Updated Review. Bosn. J. Basic Med. Sci. 2019, 20, 4320. [Google Scholar] [CrossRef]
- Mitrokhin, V.M.; Shim, A.L.; Aksyonov, A.A.; Zotov, A.S.; Konev, A.V.; Ovchinnikov, R.S.; Mladenov, I.M.; Kamkin, G.A. Circulating Interleukin-18: Association with IL-8, IL-10 and VEGF Serum Levels in Patients with and without Heart Rhythm Disorders. Int. J. Cardiol. 2016, 215, 105–109. [Google Scholar] [CrossRef]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; Farkouh, M.E.; Hsue, P.Y.; Fuster, V.; Hasan, A.A.; Amar, S. Inflammation, Immunity, and Infection in Atherothrombosis. J. Am. Coll. Cardiol. 2018, 72, 2071–2081. [Google Scholar] [CrossRef]
- Yu, Z.-M.; Deng, X.-T.; Qi, R.-M.; Xiao, L.-Y.; Yang, C.-Q.; Gong, T. Mechanism of Chronic Stress-Induced Reduced Atherosclerotic Medial Area and Increased Plaque Instability in Rabbit Models of Chronic Stress. Chin. Med. J. 2018, 131, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Zhao, Y.; Chen, A.; Lin, A. The Importance of Physiologic Ischemia Training in Preventing the Development of Atherosclerosis: The Role of Endothelial Progenitor Cells in Atherosclerotic Rabbits. Coron. Artery Dis. 2019, 30, 377–383. [Google Scholar] [CrossRef]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the Atherosclerotic Plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Wang, M.; Sui, J.; Wang, S.; Wang, X. Correlations of Carotid Intima-Media Thickness with Endothelial Function and Atherosclerosis Degree in Patients with Type 2 Diabetes Mellitus. Clin. Hemorheol. Microcirc. 2019, 72, 431–439. [Google Scholar] [CrossRef]
- Pauli, N.; Kuligowska, A.; Krzystolik, A.; Dziedziejko, V.; Safranow, K.; Rać, M.; Chlubek, D.; Rać, M.E. The Circulating Vascular Endothelial Growth Factor Is Only Marginally Associated with an Increased Risk for Atherosclerosis. Minerva Cardioangiol. 2020, 68, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, J. Role of Alternative Splicing of VEGF-A in the Development of Atherosclerosis. Aging 2018, 10, 2695–2708. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Xu, D.; Chen, W.; Chen, J.; Chen, W.; Ye, J.; Gan, S.; Zhou, W.; Song, X.; Shi, L.; et al. USP39 Promotes Malignant Proliferation and Angiogenesis of Renal Cell Carcinoma by Inhibiting VEGF-A165b Alternative Splicing via Regulating SRSF1 and SRPK1. Cancer Cell Int. 2021, 21, 486. [Google Scholar] [CrossRef]
- Kai, H.; Wu, Q.; Yin, R.; Tang, X.; Shi, H.; Wang, T.; Zhang, M.; Pan, C. LncRNA NORAD Promotes Vascular Endothelial Cell Injury and Atherosclerosis Through Suppressing VEGF Gene Transcription via Enhancing H3K9 Deacetylation by Recruiting HDAC6. Front. Cell Dev. Biol. 2021, 9, 701628. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mao, W.; Wang, L.; Lu, L.; Pang, Y. Circular RNA CircLMF1 Regulates PDGF-BB-Induced Proliferation and Migration of Human Aortic Smooth Muscle Cells by Regulating the MiR-125a-3p/VEGFA or FGF1 Axis. Clin. Hemorheol. Microcirc. 2021, Preprint, 1–17. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten Years of Anti-Vascular Endothelial Growth Factor Therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Chen, T.; Ding, Z.; Wang, Y.; Wei, Y.; Wei, X. Inhibition of FGF-FGFR and VEGF-VEGFR Signalling in Cancer Treatment. Cell Prolif. 2021, 54, e13009. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.; Bomze, D.; Dankner, R.; Fourey, D.; Boursi, B.; Arad, M.; Maor, E. Cardiovascular Toxicities of Antiangiogenic Tyrosine Kinase Inhibitors: A Retrospective, Pharmacovigilance Study. Target Oncol. 2021, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Herrmann, J. Cardiotoxicity with Vascular Endothelial Growth Factor Inhibitor Therapy. NPJ Precis. Oncol. 2018, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Rufaihah, A.J.; Johari, N.A.; Vaibavi, S.R.; Plotkin, M.; Di Thien, D.T.; Kofidis, T.; Seliktar, D. Dual Delivery of VEGF and ANG-1 in Ischemic Hearts Using an Injectable Hydrogel. Acta Biomater. 2017, 48, 58–67. [Google Scholar] [CrossRef]
- Spiller, K.L.; Koh, T.J. Macrophage-Based Therapeutic Strategies in Regenerative Medicine. Adv. Drug Deliv. Rev. 2017, 122, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Zhang, D.; Lu, L.; Qiu, H.; Wang, J. Vascular Endothelial Growth Factor-modified Macrophages Accelerate Reendothelialization and Attenuate Neointima Formation after Arterial Injury in Atherosclerosis-prone Mice. J. Cell Biochem. 2019, 120, 10652–10661. [Google Scholar] [CrossRef]
- Ceresnakova, M.; Murray, D.; Soulimane, T.; Hudson, S.P. Candidates for Smart Cardiovascular Medical Device Coatings: A Comparative Study with Endothelial and Smooth Muscle Cells. Eur. J. Pharmacol. 2021, 910, 174490. [Google Scholar] [CrossRef]
- Zhu, X.; Xie, H.; Liang, X.; Li, X.; Duan, J.; Chen, Y.; Yang, Z.; Liu, C.; Wang, C.; Zhang, H.; et al. Bilayered Nanoparticles with Sequential Release of VEGF Gene and Paclitaxel for Restenosis Inhibition in Atherosclerosis. ACS Appl. Mater. Interfaces 2017, 9, 27522–27532. [Google Scholar] [CrossRef]
- Pan, X.; Hou, R.; Ma, A.; Wang, T.; Wu, M.; Zhu, X.; Yang, S.; Xiao, X. Atorvastatin Upregulates the Expression of MiR-126 in Apolipoprotein E-Knockout Mice with Carotid Atherosclerotic Plaque. Cell. Mol. Neurobiol. 2017, 37, 29–36. [Google Scholar] [CrossRef]
- Zhao, D.; Shao, H. Effect of Blood Purification on Serum MiR-126 and VEGF Levels in the Process of Atherosclerosis in Uremic Patients under Maintenance Hemodialysis. Kaohsiung J. Med. Sci. 2018, 34, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, V.A.; Di Minno, A.; Songia, P.; Massaiu, I.; Alfieri, V.; Valerio, V.; Moschetta, D.; Andreini, D.; Alamanni, F.; Pepi, M.; et al. Sex-Specific Differences in Age-Related Aortic Valve Calcium Load: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2020, 61, 101077. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.; Chen, J.-B.; Moi, S.-H.; Yang, C.-H. Association of Proportion of the HDL-Cholesterol Subclasses HDL-2b and HDL-3 and Macrovascular Events among Patients Undergoing Hemodialysis. Sci. Rep. 2021, 11, 1871. [Google Scholar] [CrossRef]
- Milasan, A.; Jean, G.; Dallaire, F.; Tardif, J.; Merhi, Y.; Sorci-Thomas, M.; Martel, C. Apolipoprotein A-I Modulates Atherosclerosis Through Lymphatic Vessel-Dependent Mechanisms in Mice. J. Am. Hear. Assoc. 2017, 6, e006892. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Lemnitzer, P.; Gall, J.; Schwager, S.; Toska, A.; Yvan-Charvet, L.; Detmar, M.; Soehnlein, O. Arterial Delivery of VEGF-C Stabilises Atherosclerotic Lesions. Circ. Res. 2021, 128, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Gomes de Almeida Schirmer, B.; Crucet, M.; Stivala, S.; Vucicevic, G.; da Silva Barcelos, L.; Vanhoutte, P.M.; Pellegrini, G.; Camici, G.G.; Seebeck, P.; Pfundstein, S.; et al. The NO-Donor MPC-1011 Stimulates Angiogenesis and Arteriogenesis and Improves Hindlimb Ischemia via a CGMP-Dependent Pathway Involving VEGF and SDF-1α. Atherosclerosis 2020, 304, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Firnhaber, J.M.; Powell, C.S. Lower Extremity Peripheral Artery Disease: Diagnosis and Treatment. Am. Fam. Physician 2019, 99, 362–369. [Google Scholar]
- Zhang, Y.; Ren, P.; Kang, Q.; Liu, W.; Li, S.; Li, P.; Liu, H.; Shang, J.; Zhang, L.; Gong, Y.; et al. Effect of Tetramethylpyrazine on Atherosclerosis and SCAP/SREBP-1c Signaling Pathway in ApoE-/- Mice Fed with a High-Fat Diet. Evid. Based Complement. Altern. Med. 2017, 2017, 3121989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Jiao, Y.; Xie, M. Paeoniflorin Ameliorates Atherosclerosis by Suppressing TLR4-Mediated NF-ΚB Activation. Inflammation 2017, 40, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Shi, W.; Xin, Q.; Yang, B.; Hoi, M.P.; Lee, S.M.; Cong, W.; Chen, K. Tetramethylpyrazine and Paeoniflorin Inhibit Oxidized LDL-Induced Angiogenesis in Human Umbilical Vein Endothelial Cells via VEGF and Notch Pathways. Evid. Based Complement. Altern. Med. 2018, 2018, 3082507. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Mishra, A.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Wiciński, M.; Malinowski, B.; Węclewicz, M.M.; Grześk, E.; Grześk, G. Anti-Atherogenic Properties of Resveratrol: 4-Week Resveratrol Administration Associated with Serum Concentrations of SIRT1, Adiponectin, S100A8/A9 and VSMCs Contractility in a Rat Model. Exp. Ther. Med. 2017, 13, 2071–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueira, L.; González, J.C. Efecto del resveratrol sobre las concentraciones séricas del factor de crecimiento endotelial vascular durante la aterosclerosis. Clín. Investig. Arterioscler. 2018, 30, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Yang, T. Triptolide Inhibits the Progression of Atherosclerosis in Apolipoprotein E−/− Mice. Exp. Ther. Med. 2016, 12, 2307–2313. [Google Scholar] [CrossRef] [Green Version]
- Ziaei, S.; Halaby, R. Immunosuppressive, Anti-Inflammatory and Anti-Cancer Properties of Triptolide: A Mini Review. Avicenna J. Phytomed. 2016, 6, 149–164. [Google Scholar]
- Huang, Y.; Tang, S.; Ji-yan, C.; Huang, C.; Li, J.; Cai, A.; Feng, Y. Circulating MiR-92a Expression Level in Patients with Essential Hypertension: A Potential Marker of Atherosclerosis. J. Hum. Hypertens. 2017, 31, 200–205. [Google Scholar] [CrossRef]
- Yang, L.; Li, T. LncRNA TUG1 Regulates ApoM to Promote Atherosclerosis Progression through MiR-92a/FXR1 Axis. J. Cell Mol. Med. 2020, 24, 8836–8848. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tian, L.; Zhang, Z. Triptolide Inhibits Angiogenesis in Microvascular Endothelial Cells through Regulation of MiR-92a. J. Physiol. Biochem. 2019, 75, 573–583. [Google Scholar] [CrossRef]
- Marino, M.; Bo’, C.D.; Tucci, M.; Klimis-Zacas, D.; Riso, P.; Porrini, M. Modulation of Adhesion Process, E-Selectin and VEGF Production by Anthocyanins and Their Metabolites in an In Vitro Model of Atherosclerosis. Nutrients 2020, 12, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, E.F.; Zhang, Q.; Raheem, K.S.; O’Hagan, D.; O’Connell, M.A.; Kay, C.D. Common Phenolic Metabolites of Flavonoids, but Not Their Unmetabolized Precursors, Reduce the Secretion of Vascular Cellular Adhesion Molecules by Human Endothelial Cells. J. Nutr. 2016, 146, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Moral, N.; Needs, P.W.; Moyle, C.W.A.; Kroon, P.A. Hydrophobic Interactions Drive Binding between Vascular Endothelial Growth Factor-A (VEGFA) and Polyphenolic Inhibitors. Molecules 2019, 24, 2785. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931. https://doi.org/10.3390/ijms23020931
Dabravolski SA, Khotina VA, Omelchenko AV, Kalmykov VA, Orekhov AN. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. International Journal of Molecular Sciences. 2022; 23(2):931. https://doi.org/10.3390/ijms23020931
Chicago/Turabian StyleDabravolski, Siarhei A., Victoria A. Khotina, Andrey V. Omelchenko, Vladislav A. Kalmykov, and Alexander N. Orekhov. 2022. "The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets" International Journal of Molecular Sciences 23, no. 2: 931. https://doi.org/10.3390/ijms23020931
APA StyleDabravolski, S. A., Khotina, V. A., Omelchenko, A. V., Kalmykov, V. A., & Orekhov, A. N. (2022). The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. International Journal of Molecular Sciences, 23(2), 931. https://doi.org/10.3390/ijms23020931