
Deranged Myocardial Fatty Acid Metabolism in Heart Failure


{kind=link}
{kind=link}
Abstract
:1. Introduction
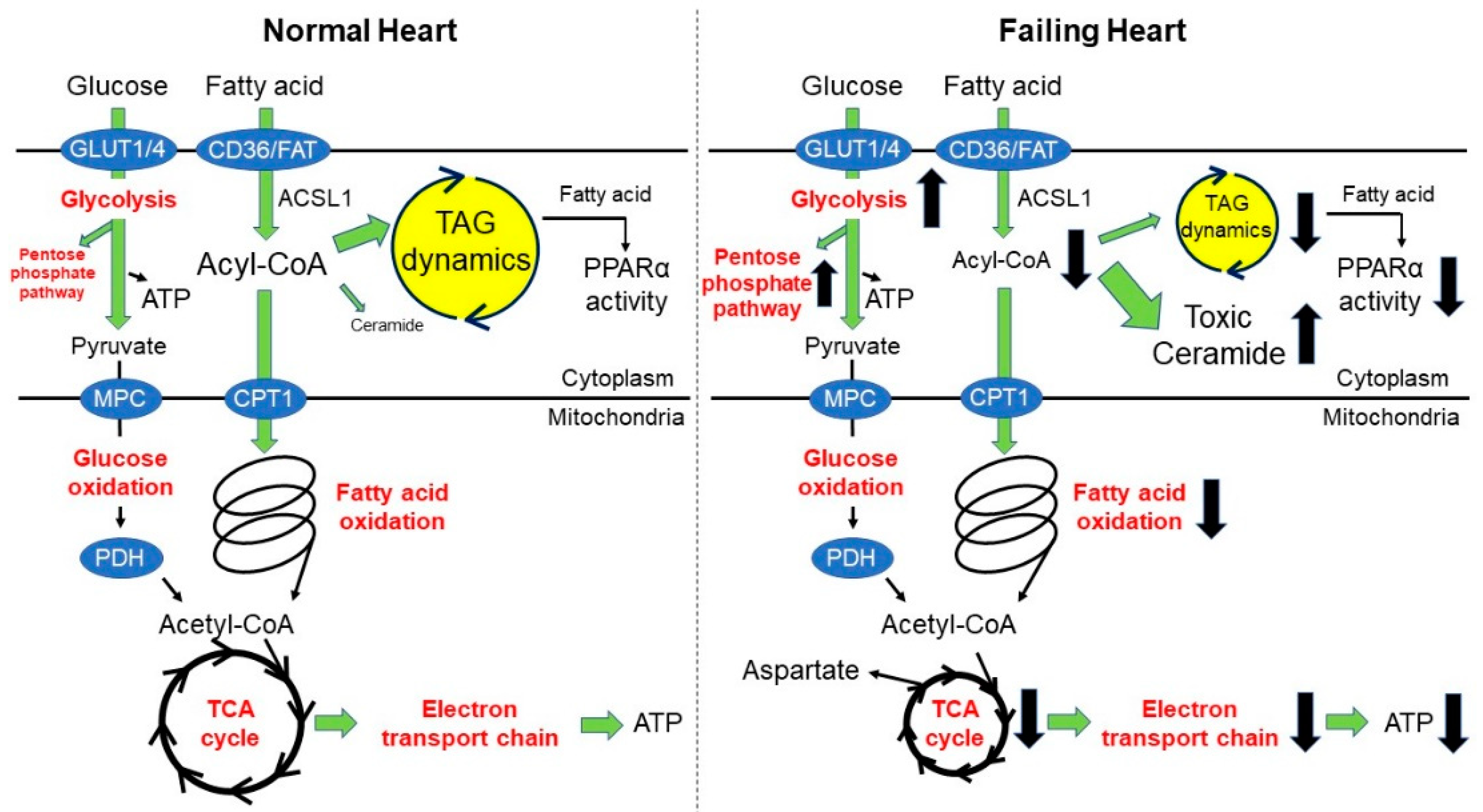
2. Fatty Acid Oxidation and Heart Failure
2.1. What Do We Know in This Area?
2.2. Recent Advances in This Area
3. Triacylglyceride Dynamics and Heart Failure
3.1. What Do We Know in This Area?
3.2. Recent Advances in This Area
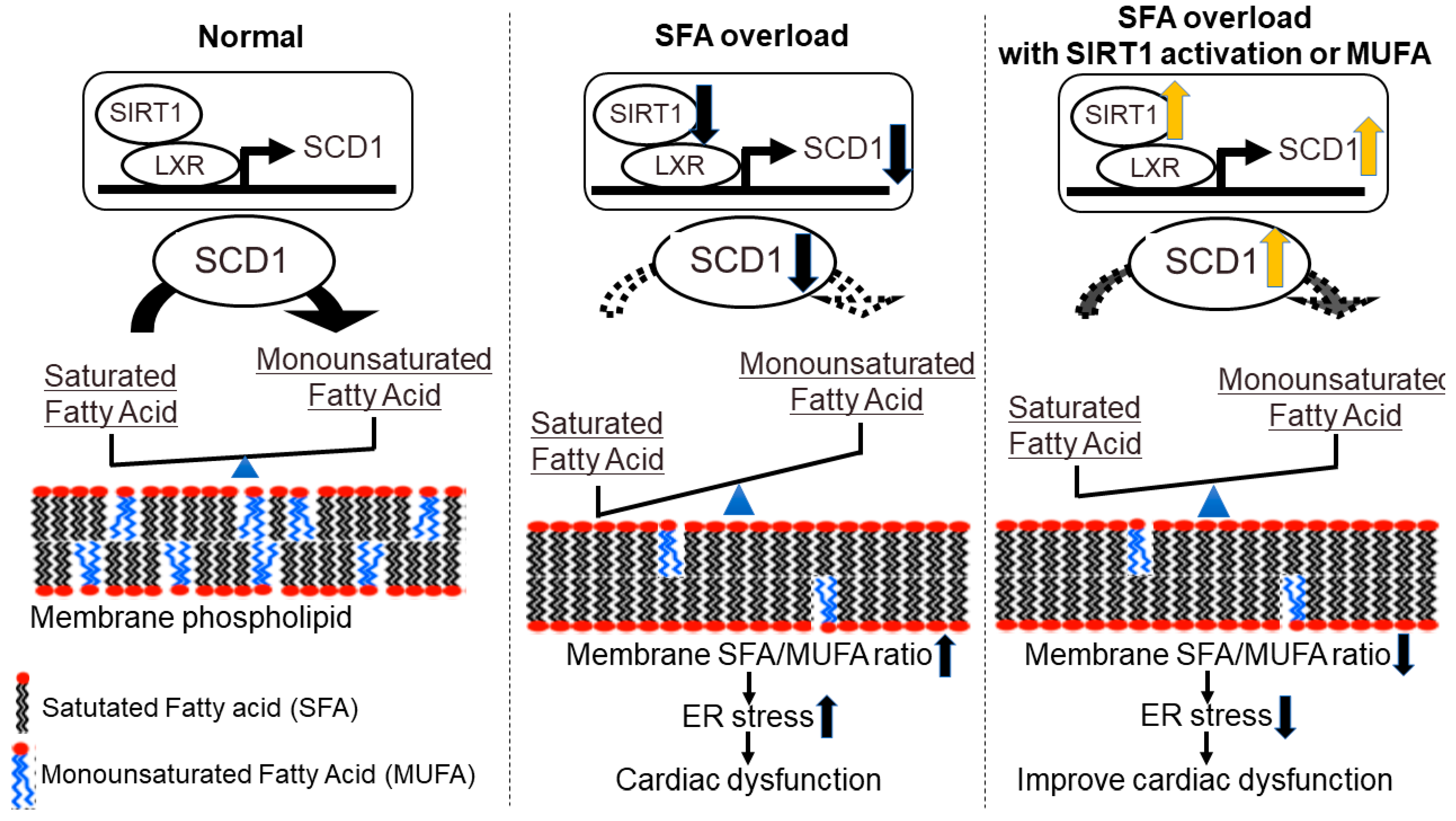
4. Membrane Fatty Acid Composition and Heart Failure-New Concept Targeting Lipid Metabolism in Heart Failure
5. Developing Heart Failure Therapy by Nutritional and Pharmacological Intervention
5.1. Statin and Fibrate Therapy
5.2. Ketone Body Supplementation and SGLT2 Inhibitors
5.3. Short-to Medium-Chain Fatty Acid Supplementation
5.4. MUFA and PUFA Supplementation
5.5. Boosting NAD+ Level by Supplementation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saddik, M.; Lopaschuk, G.D. Myocardial Triglyceride Turnover and Contribution to Energy Substrate Utilization in Isolated Working Rat Hearts. J. Biol. Chem. 1991, 266, 8162–8170. [Google Scholar] [CrossRef]
- Wisneski, J.A.; Stanley, W.C.; Neese, R.A.; Gertz, E.W. Effects of Acute Hyperglycemia on Myocardial Glycolytic Activity in Humans. J. Clin. Investig. 1990, 85, 1648–1656. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive Quantification of Fuel Use by the Failing and Nonfailing Human Heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Szczepaniak, L.S.; Dobbins, R.L.; Metzger, G.J.; Sartoni-D’Ambrosia, G.; Arbique, D.; Vongpatanasin, W.; Unger, R.; Victor, R.G. Myocardial Triglycerides and Systolic Function in Humans: In Vivo Evaluation by Localized Proton Spectroscopy and Cardiac Imaging. Magn. Reson Med. 2003, 49, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Herrero, P.; Peterson, L.R.; McGill, J.B.; Mattew, S.; Lesniak, D.; Dence, C.; Gropler, R.J. Increased Myocardial Fatty Acid Metabolism in Patients with Type 1 Diabetes Mellitus. J. Am. Coll. Cardiol. 2006, 47, 598–604. [Google Scholar] [CrossRef]
- McGavock, J.M.; Lingvay, I.; Zib, I.; Tillery, T.; Salas, N.; Unger, R.; Levine, B.D.; Raskin, P.; Victor, R.G.; Szczepaniak, L.S. Cardiac Steatosis in Diabetes Mellitus: A 1H-Magnetic Resonance Spectroscopy Study. Circulation 2007, 116, 1170–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutger, V.W.; Luuk, J.R.; Michaela, D.; Sebastiaan, H.; Michael, S.; Jeroen, J.B.; Johannes, W.A.; Johannes, A.R.; Albert, D.R.; Hildo, J.L. The Ageing Male Heart: Myocardial Triglyceride Content as Independent Predictor of Diastolic Function. Eur. Heart J. 2008, 29, 1516–1522. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic Heart Disease in Obese Rats: Implications for Human Obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [Green Version]
- Schaffeur, J.E. Lipotoxicity: When Tissues Overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef]
- Yagyu, H.; Chen, G.; Yokoyama, M.; Hirata, K.; Augustus, A.; Kako, Y.; Seo, T.; Hu, Y.; Lutz, P.E.; Merkel, M.; et al. Lipoprotein Lipase (Lpl) on the Surface of Cardiomyocytes Increases Lipid Uptake and Produces a Cardiomyopathy. J. Clin. Investig. 2003, 111, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Kovacs, A.; Blanton, R.M.; Han, X.; Courtois, M.; Weinheimer, C.J.; Yamada, K.A.; Brunet, S.B.; Xu, H.; Nerbonne, J.M.; et al. Transgenic Expression of Fatty Acid Transport Protein 1 in the Heart Causes Lipotoxic Cardiomyopathy. Circ. Res. 2005, 96, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The Cardiac Phenotype Induced by Pparalpha Overexpression Mimics That Caused by Diabetes Mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Yang, J.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Kovacs, A.; Febbraio, M.; Finck, B.N.; Kelly, D.P.K. CD36 Deficiency Rescues Lipotoxic Cardiomyopathy. Circ. Res. 2007, 100, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.G.; Bharadwaj, K.G.; Fong, J.L.; Mitra, R.; Sambandam, N.; Courtois, M.R.; Lavine, K.J.; Goldberg, I.J.; Kelly, D.P. Rescue of Cardiomyopathy in Peroxisome Proliferator-Activated Receptor-Alpha Transgenic Mice by Deletion of Lipoprotein Lipase Identifies Sources of Cardiac Lipids and Peroxisome Proliferator-Activated Receptor-Alpha Activators. Circulation 2010, 121, 426–435. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Overturf, M.L. Effects of Moderate Hypertension on Cardiac Function and Metabolism in the Rabbit. Hypertension 1988, 11, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty Acid Oxidation Enzyme Gene Expression is Downregulated in The Failing Heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Olson, D.P.; Marley, L.C.; Garcia-Menendez, L.; Synovec, R.E.; Tian, R. Cardiac-Specific Deletion of Acetyl Coa Carboxylase 2 Prevents Metabolic Remodeling During Pressure-Overload Hypertrophy. Circ. Res. 2012, 111, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.J.; Garland, P.B.; Hakes, C.N.; Newsholme, E.A. The Glucose Fatty-Acid Cycle. Its Role in Insulin Sensitivity and the Metabolic Disturbances of Diabetes Mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Tornheim, K.; Leahy, J.L. Glucose-Fatty Acid Cycle to Inhibit Glucose Utilization and Oxidation is Not Operative in Fatty Acid-Cultured Islets. Diabetes 1999, 48, 1747–1753. [Google Scholar] [CrossRef]
- Thompson, L.; Cooney, G.J. Acyl-CoA Inhibition of Hexokinase in Rat and Human Skeletal Muscle is A Potential Mechanism of Lipid-Induced Insulin Resistance. Diabetes 2000, 49, 1761. [Google Scholar] [CrossRef]
- Laura, J.M.; Claudia, N.M.A.; Lisa, C.H. Positioning Metabolism as a Central Player in the Diabetic Heart. J. Lipid. Atheroscler. 2020, 9, 92–109. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. the Randle Cycle Revisited: A New Head for an Old Hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. The Pathogenesis of Insulin Resistance: Integrating Signaling Pathways and Substrate Flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosatos, K.; Pollak, N.M.; Pol, C.J.; Ntziachristos, P.; Willecke, F.; Valenti, M.C.; Trent, C.M.; Hu, Y.; Guo, S.; Aifantis, I.; et al. Cardiac Myocyte KLF5 Regulates Ppara Expression and Cardiac Function. Circ. Res. 2016, 118, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Augustus, S.A.; Buchanan, J.; Park, T.k.; Hirata, K.; Noh, H.L.; Sun, J.; Homma, S.; D’armiento, J.; Abel, E.D.; Goldberg, I.J. Loss of Lipoprotein Lipase-Derived Fatty Acids Leads to Increased Cardiac Glucose Metabolism and Heart Dysfunction. J. Biol. Chem. 2006, 281, 8716–8723. [Google Scholar] [CrossRef] [Green Version]
- Umbarawan, Y.; Syamsunarno, M.R.A.A.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sano, M.; Sunaga, H.; et al. Myocardial Fatty Acid Uptake Through Cd36 is Indispensable for Sufficient Bioenergetic Metabolism to Prevent Progression of Pressure Overload-Induced Heart Failure. Sci. Rep. 2018, 8, 12035. [Google Scholar] [CrossRef] [PubMed]
- Umbarawan, Y.; Syamsunarno, M.R.A.A.; Koitabashi, N.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Nagahata-Naito, Y.; Obinata, H.; Sano, M.; Sunaga, H.; et al. Glucose is Preferentially Utilized for Biomass Synthesis in Pressure-Overloaded Hearts: Evidence From Fatty Acid-Binding Protein-4 and -5 Knockout Mice. Cardiovasc. Res. 2018, 114, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Rees, M.L.; Subramaniam, J.; Li, Y.; Hamilton, D.J.; Frazier, O.H.; Taegtmeyer, H.A. A Pkm2 Signature in The Failing Heart. Biochem. Biophys. Res. Commun. 2015, 459, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, S.; Fujita, J.; Hishiki, T.; Matsuura, T.; Hattori, F.; Ohno, R.; Kanazawa, H.; Seki, T.; Nakajima, K.; Kishino, Y.; et al. Glutamine Oxidation is Indispensable for Survival of Human Pluripotent Stem Cells. Cell. Metab. 2016, 23, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritterhoff, J.; Young, S.; Villet, O.; Shao, D.; Neto, F.C.; Bettcher, L.F.; Hsu, Y.A.; Kolwicz, S.C., Jr.; Raftery, D.; Tian, R. Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis. Circ. Res. 2019, 126, 182–196. [Google Scholar] [CrossRef]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2016, 6, 331–351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jaswal, J.S.; Ussher, J.R.; Sankaralingam, S.; Wagg, C.; Zaugg, M.; Lopaschuk, G.D. Cardiac Insulin-Resistance and Decreased Mitochondrial Energy Production Precede the Development of Systolic Heart Failure After Pressure-Overload Hypertrophy. Circ. Heart Fail 2013, 6, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Zhabyeyev, P.; Gandhi, M.; Mori, J.; Basu, R.; Kassiri, Z.; Clanachan, A.; Lopaschuk, G.D.; Oudit, G.Y. Pressure-Overload-Induced Heart Failure Induces a Selective Reduction in Glucose Oxidation at Physiological Afterload. Cardiovasc. Res. 2013, 97, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, M.A.; Lau, A.Z.; Chen, A.P.; Gu, Y.; Nagendran, J.; Barry, J.; Hu, X.; Dyck, J.R.; Tyler, D.J.; Clarke, K.; et al. Hyperpolarized (13)C Magnetic Resonance Reveals Early- and Late-Onset Changes to in Vivo Pyruvate Metabolism in The Failing Heart. Eur. J. Heart Fail 2013, 15, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recchia, F.A.; McConnell, P.I.; Bernstein, R.D.; Vogel, T.R.; Xu, X.; Hintze, T.H. Reduced Nitric Oxide Production and Altered Myocardial Metabolism During the Decompensation of Pacing-Induced Heart Failure in the Conscious Dog. Circ. Res. 1998, 83, 969–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Matsuhashi, T.; Hishiki, T.; Zhou, H.; Ono, T.; Kaneda, R.; Iso, T.; Yamaguchi, A.; Endo, J.; Katsumata, Y.; Atsushi, A.; et al. Activation of Pyruvate Dehydrogenase by Dichloroacetate Has the Potential to Induce Epigenetic Remodeling in the Heart. J. Mol. Cell. Cardiol. 2015, 82, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of A Member of the Steroid Hormone Receptor Superfamily by Peroxisome Proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Djouadi, F.; Brandt, J.M.; Weinheimer, C.J.; Leone, T.C.; Gonzalez, F.J.; Kelly, D.P. the Role of the Peroxisome Proliferator-Activated Receptor Alpha (Ppar Alpha) in The Control of Cardiac Lipid Metabolism. Prostaglandins Leukot. Essent. Fat Acids 1999, 60, 339–343. [Google Scholar] [CrossRef]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A Critical Role for the Peroxisome Proliferator-Activated Receptor Alpha (Pparalpha) in The Cellular Fasting Response: The PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Fujii, H.; Takahashi, T.; Kodama, M.; Aizawa, Y.; Ohta, Y.; Ono, T.; Hasegawa, G.; Naito, M.; Nakajima, T.; et al. Constitutive Regulation of Cardiac Fatty Acid Metabolism Through Peroxisome Proliferator-Activated Receptor Alpha Associated with Age-Dependent Cardiac Toxicity. J. Biol. Chem. 2000, 275, 22293–22299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguere, V.; Kelly, D.P. Estrogen-Related Receptor Alpha Directs Peroxisome Proliferator-Activated Receptor Alpha Signaling in the Transcriptional Control of Energy Metabolism in Cardiac and Skeletal Muscle. Mol. Cell. Biol. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, T.; Matsuura, R.T.; Wan, S.; Ryba, M.D.; Kim, U.J.; Won, J.K.; Lai, L.; Petucci, C.; Petrenko, N.; Musunuru, K.; et al. A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ. Res. 2020, 126, 1685–1702. [Google Scholar] [CrossRef]
- Kwon, D.H.; Eom, G.H.; Kee, H.J.; Nam, Y.S.; Cho, Y.K.; Kim, D.K.; Koo, J.Y.; Kim, H.S.; Nam, K.l.; Kim, K.K.; et al. Estrogen-Related Receptor Gamma Induces Cardiac Hypertrophy by Activating GATA4. J. Mol. Cell. Cardiol. 2013, 65, 88–97. [Google Scholar] [CrossRef]
- Lasheras, J.; Pardon, R.; Velilla, M.; Poncelas, M.; Salvatella, N.; Simo, R.; Ruiz-Meana, M.; Zamora, M.; Villena, J.A. Cardiac-Specific Overexpression of ERRγ in Mice Induces Severe Heart Dysfunction and Early Lethality. Int. J. Mol. Sci. 2021, 22, 8047. [Google Scholar] [CrossRef] [PubMed]
- Prosdocimo, D.A.; John, J.E.; Zhang, L.; Efraim, S.E.; Zhang, R.; Liao, X.; Jain, M.K. KLF15 and PPARα Cooperate to Regulate Cardiomyocyte Lipid Gene Expression and Oxidation. PPAR Res. 2015, 2015, 201625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosdocimo, D.A.; Anand, P.; Liao, X.; Zhu, H.; Shelkay, S.; Artero-Calderon, P.; Zhang, L.; Kirsh, J.; Moore, D.; Rosca, M.G.; et al. Kruppel-Like Factor 15 is A Critical Regulator of Cardiac Lipid Metabolism. J. Biol. Chem. 2014, 289, 5914–5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugi, K.; Hsieh, P.N.; Ilkayeva, O.; Shelkay, S.; Moroney, B.; Baadh, P.; Haynes, B.; Pophal, M.; Fan, L.; Newgard, C.B.; et al. Kruppel-Like Factor 15 is Required for the Cardiac Adaptive Response to Fasting. PLoS ONE 2018, 13, e0192376. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Zhang, R.; Lu, Y.; Prosdocimo, D.A.; Sangwung, P.; Zhang, L.; Zhou, G.; Anand, P.; Lai, L.; Leone, T.C.; et al. Kruppel-Like Factor 4 is Critical for Transcriptional Control of Cardiac Mitochondrial Homeostasis. J. Clin. Investig. 2015, 125, 3461–3476. [Google Scholar] [CrossRef] [Green Version]
- Vega, R.B.; Kelly, D.P. Cardiac Nuclear Receptors: Architects of Mitochondrial Structure and Function. J. Clin. Investig. 2017, 127, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Promotes Cardiac Mitochondrial Biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, L.K.; Mansfield, C.M.; Lehman, J.J.; Kovacs, A.; Courtois, M.; Saffitz, J.E.; Medeiros, D.M.; Valencik, M.L.; McDonald, J.A.; Kelly, D.P. Cardiac-Specific Induction of the Transcriptional Coactivator Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1alpha Promotes Mitochondrial Biogenesis and Reversible Cardiomyopathy in A Developmental Stage-Dependent Manner. Circ. Res. 2004, 94, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, J.J.; Boudina, S.; Banke, N.H.; Sambandam, N.; Han, X.; Young, D.M.; Leone, T.C.; Gross, R.W.; Lewandowski, E.D.; Abel, E.D.; et al. The Transcriptional Coactivator Pgc-1alpha is Essential for Maximal and Efficient Cardiac Mitochondrial Fatty Acid Oxidation and Lipid Homeostasis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H185–H196. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse Aortic Constriction Leads to Accelerated Heart Failure in Mice Lacking Ppar-Gamma Coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.P.; Kao, C.Y.; Wang, L.; Creighton, C.J.; Yang, J.; Donti, T.R.; Harmancey, R.; Vasquez, H.G.; Graham, B.H.; Bellen, H.J.; et al. Increased COUP-TFII Expression in Adult Hearts Induces Mitochondrial Dysfunction Resulting in Heart Failure. Nat. Commun. 2015, 6, 8245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritah, A.; Steel, J.H.; Nichol, D.; Parker, N.; Williams, S.; Price, A.; Strauss, L.; Ryder, T.A.; Mobberley, M.A.; Poutanen, M.; et al. Elevated Expression of the Metabolic Regulator Receptor-Interacting Protein 140 Results in Cardiac Hypertrophy and Impaired Cardiac Function. Cardiovasc. Res. 2010, 86, 443–451. [Google Scholar] [CrossRef]
- Fernandez-Caggiano, M.; Kamynina, A.; Francois, A.A.; Prysyazhna, O.; Eykyn, T.R.; Krasemann, S.; Crespo-Leiro, M.G.; Vieites, M.G.; Bianchi, K.; Morales, V.; et al. Mitochondrial Pyruvate Carrier Abundance Mediates Pathological Cardiac Hypertrophy. Nat. Metab. 2020, 2, 1223–1231. [Google Scholar] [CrossRef]
- McCommis, K.S.; Kovacs, A.; Weinheimer, C.J.; Shew, T.M.; Koves, T.R.; Ilkayeva, O.R.; Kamm, D.R.; Pyles, K.D.; King, M.T.; Veech, R.L.; et al. Nutritional Modulation of Heart Failure in Mitochondrial Pyruvate Carrier-Deficient Mice. Nat. Metab. 2020, 2, 1232–1247. [Google Scholar] [CrossRef]
- Zhang, Y.; Taufalele, P.V.; Cochran, J.D.; Robillard-Frayne, I.; Marx, J.; Soto, J.; Rauckhorst, A.J.; Tayyari, F.; Pewa, A.D.; Gray, L.R.; et al. Mitochondrial Pyruvate Carriers Are Required for Myocardial Stress Adaptation. Nat. Metab. 2020, 2, 1248–1264. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Badolia, R.B.; Lettlova, S.; Parnell, K.M.; Shankar, T.S.; Diakos, N.A.; Olson, K.A.; Taleb, I.; Tatum, S.M.; Berg, J.A.; et al. the Pyruvate-Lactate Axis Modulates Cardiac Hypertrophy and Heart Failure. Cell. Metab. 2021, 33, 629–648. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, N.; O’Donnell, J.M.; McKinney, R.D.; Pound, K.M.; Woldegiorgis, G.; LaNoue, K.F.; Ballal, K.; Taegtmeyer, H.; Buttrick, P.M.; Lewandowski, E.D. Recruitment of Compensatory Pathways to Sustain Oxidative Flux with Reduced Carnitine Palmitoyltransferase I Activity Characterizes Inefficiency in Energy Metabolism in Hypertrophied Hearts. Circulation 2007, 115, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Sansbury, B.E.; DeMartino, A.M.; Xie, Z.; Brooks, A.C.; Brainard, R.E.; Watson, L.J.; DeFilippis, A.P.; Cummins, T.D.; Harbeson, M.A.; Brittian, K.R.; et al. Metabolomic Analysis of Pressure-Overloaded and Infarcted Mouse Hearts. Circ. Heart Fail. 2014, 7, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, D.; Kolwicz, S.C., Jr.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation 2020, 142, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Liu, T.; Husain, S.; Zhai, P.; Warren, J.S.; Hsu, C.P.; Matsuda, T.; Phiel, C.J.; Cox, J.E.; Tian, B.; et al. Glycogen Synthase Kinase-3α Promotes Fatty Acid Uptake and Lipotoxic Cardiomyopathy. Cell. Metab. 2019, 29, 1119–1134. [Google Scholar] [CrossRef]
- O’Donnell, J.M.; Fields, A.D.; Sorokina, N.; Lewandowski, E.D. The Absence of Endogenous Lipid Oxidation in Early Stage Heart Failure Exposes Limits in Lipid Storage and Turnover. J. Mol. Cell. Cardiol. 2008, 44, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.M.; Zampino, M.; Alpert, N.M.; Fasano, M.J.; Geenen, D.L.; Lewandowski, E.D. Accelerated Triacylglycerol Turnover Kinetics in Hearts of Diabetic Rats Include Evidence for Compartmented Lipid Storage. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E448–E455. [Google Scholar] [CrossRef] [Green Version]
- Shang, R.; Rodrigues, B. Lipoprotein Lipase and Its Delivery of Fatty Acids to the Heart. Biomolecules 2021, 11, 1016. [Google Scholar] [CrossRef]
- Levak-Frank, S.; Radner, H.; Walsh, A.; Stollberger, R.; Knipping, G.; Hoefler, G.; Sattler, W.; Weinstock, P.H.; Breslow, J.L.; Zechner, R. Muscle-Specific Overexpression of Lipoprotein Lipase Causes a Severe Myopathy Characterized by Proliferation of Mitochondria and Peroxisomes in Transgenic Mice. J. Clin. Investig. 1995, 96, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Pulawa, L.K.; Eckel, R.H. Overexpression of Muscle Lipoprotein Lipase and Insulin Sensitivity. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 569–574. [Google Scholar] [CrossRef]
- Noh, H.L.; Okajima, K.; Molkentin, J.D.; Homma, S.; Goldberg, I.J. Acute Lipoprotein Lipase Deletion in Adult Mice Leads to Dyslipidemia and Cardiac Dysfunction. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E755–E760. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Kim, M.S.; Puthanveetil, P.; Ghosh, S.; Luciani, D.S.; Johnson, J.D.; Abrahani, A.; Rodrigues, B. Glucose-Induced Endothelial Heparanase Secretion Requires Cortical and Stress Actin Reorganization. Cardiovasc. Res. 2010, 87, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafat, I.; Ilan, N.; Zoabi, S.; Vlodavsky, I.; Nakhoul, F. Heparanase Levels Are Elevated in the Urine and Plasma of Type 2 Diabetes Patients and Associate with Blood Glucose Levels. PLoS ONE 2011, 6, e17312. [Google Scholar] [CrossRef] [Green Version]
- Pei-Ling Chiu, A.; Wang, F.; Lal, N.; Wang, Y.; Zhang, D.; Hussein, B.; Wan, A.; Vlodavsky, I.; Rodrigues, B. Endothelial Cells Respond to Hyperglycemia by Increasing the Lpl Transporter GPIHBP1. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1274–E1283. [Google Scholar] [CrossRef] [Green Version]
- Carley, N.A.; Lewandowski, D.E. Triacylglycerol Turnover in The Failing Heart. Biochim. Biophys. Acta 2016, 1861, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, A.; Drosatos, K.; Cheema, F.H.; Ji, R.; Khawaja, T.; Yu, S.; Kato, T.; Khan, R.; Takayama, H.; Knöll, R.; et al. Ventricular Assist Device Implantation Corrects Myocardial Lipotoxicity, Reverses Insulin Resistance, and Normalizes Cardiac Metabolism in Patients with Advanced Heart Failure. Circulation 2012, 125, 2844–2853. [Google Scholar] [CrossRef] [Green Version]
- Barger, M.P.; Brandt, M.J.; Leone, C.T.; Weinheimer, J.C.; Kelly, P.D. Deactivation of Peroxisome Proliferator-Activated Receptor-Alpha During Cardiac Hypertrophic Growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-Mediated Fat Catabolism Regulates Cardiac Mitochondrial Function Via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Roe, N.D.; Handzlik, M.K.; Li, T.; Tian, R. The Role of Diacylglycerol Acyltransferase (DGAT) 1 and 2 in Cardiac Metabolism and Function. Sci. Rep. 2018, 8, 4983. [Google Scholar] [CrossRef] [Green Version]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a Lipid Droplet-Binding Protein, Protects Heart from Oxidative Burden by Sequestering Fatty Acid from Excessive Oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolwicz, S.C., Jr.; Liu, L.; Goldberg, I.J.; Tian, R. Enhancing Cardiac Triacylglycerol Metabolism Improves Recovery from Ischemic Stress. Diabetes 2015, 64, 2817–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Hannaert, C.J.; Hoorens, A.; Eizirik, L.D.; Pipeleers, G.D. Inverse Relationship Between Cytotoxicity of Free Fatty Acids in Pancreatic Islet Cell.s and Cell.ular Triglyceride Accumulation. Diabetes 2001, 50, 1771–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, J.E.; Vork, M.M.; Roemen, T.H.; de Jong, Y.F.; Cleutjens, J.P.; van der Vusse, G.J.; van Bilsen, M. Saturated but Not Mono-Unsaturated Fatty Acids Induce Apoptotic Cell. Death in Neonatal Rat Ventricular Myocytes. J. Lipid. Res. 1997, 38, 1384–1394. [Google Scholar]
- Maedler, K.; Spinas, A.G.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, Y.M. Distinct Effects of Saturated and Monounsaturated Fatty Acids on Beta-Cell. Turnover and Function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated Fatty Acids Induce C-Src Clustering Within Membrane Subdomains, Leading to Jnk Activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, K.; Bugger, H.; Wende, R.A.; Soto, J.; Jenson, A.G.; Tor, R.A.; McGlauflin, R.; Kenny, C.H.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Lahey, R.; Wang, X.; Carley, N.A.; Lewandowski, D.E. Dietary Fat Supply to Failing Hearts Determines Dynamic Lipid Signaling for Nuclear Receptor Activation and Oxidation of Stored Triglyceride. Circulation 2014, 130, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, R.J.; Carley, N.A.; Ji, R.; Zhang, X.; Fasano, M.; Schulze, C.P.; Lewandowski, D.E. Preservation of Acyl Coenzyme a Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation 2019, 139, 2765–2777. [Google Scholar] [CrossRef]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, J.P.; Brunjes, L.D.; Castillero, E.; Zhang, X.; Deng, Y.L.; et al. Increased De Novo Ceramide Synthesis and Accumulation in Failing Myocardium. JCI Insight 2017, 2, e82922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menuz, V.; Howell, K.S.; Gentina, S.; Epstein, S.; Riezman, I.; Fornallaz-Mulhauser, M.; Hengartner, M.O.; Gomez, M.; Riezman, H.; Martinou, J.C. Protection of C. elegans from Anoxia by HYL-2 Ceramide Synthase. Science 2009, 324, 381–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of Endoplasmic Reticulum Structure and Integrity in Lipotoxic Cell. Death. J. Lipid. Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, A.; Weigert, C.; Staiger, H.; Rittig, K.; Cegan, A.; Lutz, P.; Machicao, F.; Häring, H.U.; Schleicher, E. Induction of Stearoyl-Coa Desaturase Protects Human Arterial Endothelial Cells Against Lipotoxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 43, E339–E349. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Wang, D.; Pagliassotti, M.J. Saturated Fatty Acid-Mediated Endoplasmic Reticulum Stress and Apoptosis Are Augmented by Trans-10, Cis-12-Conjugated Linoleic Acid in Liver Cells. Mol. Cell. Biochem. 2007, 303, 105–113. [Google Scholar] [CrossRef]
- Ariyama, H.; Kono, N.; Matsuda, S.; Inoue, T.; Arai, H. Decrease in Membrane Phospholipid Unsaturation Induces Unfolded Protein Response. J. Biol. Chem. 2010, 285, 22027–22035. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Yoshida, N.; Isobe, S.; Moriyama, H.; Goto, S.; et al. Decrease in Membrane Phospholipids Unsaturation Correlates with Myocardial Diastolic Dysfunction. PLoS ONE 2018, 13, e0208396. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Yoshida, N.; Isobe, S.; Moriyama, H.; Goto, S.; et al. Sirt1 Counteracts Decrease in Membrane Phospholipid Unsaturation and Diastolic Dysfunction During Saturated Fatty Acid Overload. J. Mol. Cell. Cardiol. 2019, 133, 1–11. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shinmura, K. Targeting sirtuins to modulate energy metabolism in heart disease. In Sirtuin Biology in Medicine, Edition; Maiese, K., Ed.; Academic Press Elsevier: Amsterdam, The Netherlands, 2021; pp. 285–293. ISBN 9780128141182. [Google Scholar]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Katsumata, Y.; Shirakawa, K.; Isobe, S.; Moriyama, H.; Goto, S.; Shimanaka, Y.; Kono, N.; et al. Saturated Fatty Acid-Induced Cardiomyopathy with Diastolic Dysfunction Can Be Ameliorated by Changing the Quality of Fatty Acids to Monounsaturated Fatty Acid. Arch. Med. Sci. 2021, in press. [CrossRef]
- Costantino, S.; Paneni, F.; Cosentino, F. Ageing, Metabolism and Cardiovascular Disease. J. Physiol. 2016, 594, 2061–2073. [Google Scholar] [CrossRef]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Isobe, S.; Moriyama, H.; Goto, S.; Shimanaka, Y.; et al. Palmitate Induces Cardiomyocyte Death Via Inositol Requiring Enzyme-1 (IRE1)-Mediated Signaling Independent of X-Box Binding Protein 1 (XBP1). Biochem. Biophys. Res. Commun. 2020, 526, 122–127. [Google Scholar] [CrossRef]
- Balakumar, P.; Mahadevan, N. Interplay Between Statins and PPARS in Improving Cardiovascular Outcomes: A Double-Edged Sword? Br. J. Pharmacol. 2012, 165, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Sambathkumar, R.; Mahadevan, N.; Muhsinah, A.B.; Alsayari, A.; Venkateswaramurthy, N.; Dhanaraj, S.A. Molecular Targets of Fenofibrate in the Cardiovascular-Renal Axis: A unifying perspective of its pleiotropic benefits. Pharmacol. Res. 2019, 144, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Matsumura, T.; Senokuchi, T.; Ishii, N.; Murata, Y.; Taketa, K.; Motoshima, H.; Taguchi, T.; Sonoda, K.; Kukidome, D.; et al. Statins Activate Peroxisome Proliferator-Activated Receptor Gamma Through ExtraCell.ular Signal-Regulated Kinase 1/2 and p38 Mitogen-Activated Protein Kinase-Dependent Cyclooxygenase-2 Expression in Macrophages. Circ. Res. 2007, 100, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.W.; Ye, P.; He, J.Q.; Sheng, L.; Wang, L.Y.; Du, J. Simvastatin inhibited cardiac hypertrophy and fibrosis in apolipoprotein E-deficient mice fed a ‘Western-style diet’ by increasing PPARα and γ expression and reducing TC, MMP-9, and Cat S levels. Acta Pharmacol. Sin. 2010, 31, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wu, H.; Wang, C.; Shao, H.; Huang, H.; Jing, H.; Li, D. Simvastatin attenuates cardiopulmonary bypass-induced myocardial inflammatory injury in rats by activating peroxisome proliferator-activated receptor γ. Eur. J. Pharmacol. 2010, 649, 255–262. [Google Scholar] [CrossRef]
- Kaimoto, S.; Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Tateishi, S.; Ono, K.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Activation of PPAR-α in the Early Stage of Heart Failure Maintained Myocardial Function and Energetics in Pressure-Overload Heart Failure. Am. J. Physiol. Heart. Circ. Physiol. 2017, 312, H305–H313. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.D.; Paynter, N.P.; Everett, B.M.; Glynn, R.J.; Amarenco, P.; Elam, M.; Ginsberg, H.; Hiatt, W.R.; Ishibashi, S.; Koenig, W.; et al. Rationale and design of the Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study. Am. Heart J. 2018, 206, 80–93. [Google Scholar] [CrossRef]
- Lommi, J.; Kupari, M.; Koskinen, P.; Näveri, H.; Leinonen, H.; Pulkki, K.; Härkönen, M. Blood ketone bodies in congestive heart failure. J. Am. Coll. Cardiol. 1996, 28, 665–672. [Google Scholar] [CrossRef]
- Arima, Y.; Nakagawa, Y.; Takeo, T.; Ishida, T.; Yamada, T.; Hino, S.; Nakao, M.; Hanada, S.; Umemoto, T.; Suda, T.; et al. Murine Neonatal Ketogenesis Preserves Mitochondrial Energetics by Preventing Protein Hyperacetylation. Nat. Metab. 2021, 3, 196–210. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; DeMets, D.L.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Langkilde, A.M.; Martinez, F.A.; Bengtsson, O.; Ponikowski, P.; Sabatine, M.S.; et al. A Trial to Evaluate the Effect of the Sodium-Glucose Co-Transporter 2 Inhibitor Dapagliflozin on Morbidity and Mortality in Patients with Heart Failure and Reduced Left Ventricular Ejection Fraction (DAPA-HF). Eur. J. Heart Fail. 2019, 21, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Sano, M.; Goto, S. Possible Mechanism of Hematocrit Elevation by Sodium Glucose Cotransporter 2 Inhibitors and Associated Beneficial Renal and Cardiovascular Effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef]
- Yurista, S.R.; Chong, C.R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic Potential of Ketone Bodies for Patients with Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell. Metab. 2020, 32, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Odanovic, N.; Nakada, Y.; Dohi, S.; Zhai, P.; Ivessa, A.; Yang, Z.; Abdellatif, M.; Sadoshima, J. Dietary Carbohydrates Restriction Inhibits the Development of Cardiac Hypertrophy and Heart failure. Cardiovasc. Res. 2021, 117, 2365–2376. [Google Scholar] [CrossRef]
- Cheng, C.; Biton, M.; Haber, L.A.; Gunduz, N.; Eng, G.; Gaynor, T.L.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, L.V.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell. Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, D.M.; Newman, J.; He, W.; Shirakawa, K.; Moan, L.N.; Grueter, A.C.; Lim, H.; Saunders, R.L.; Stevens, D.R.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, E.D.; Raymond, K.K.; Lawrence, T.W.; O’Donnell, J.M.; Stephen, F.V. Mitochondrial Preference for Short Chain Fatty Acid Oxidation During Coronary Artery Constriction. Circulation 2002, 105, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Zhang, B.; Suzuki, A.; Yamaguchi, S.; Adachi, J.; Tomonaga, T.; Yasunaga, S.; Saku, K.; Aoyama, T.; Hirano, K. Effect of Tricaprin on Cardiac Proteome in a Mouse Model for Triglyceride Deposit Cardiomyovasculopathy. J. Oleo Sci. 2020, 69, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, P.; Zhang, Y.; He, W.; Chen, X.; Chen, J.; He, L.; Mao, L.; Wu, F.; Jiao, J. Dietary Fats in Relation to Total and Cause-Specific Mortality in a Prospective Cohort of 521 120 Individuals with 16 Years of Follow-Up. Circ. Res. 2019, 124, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Carbone, S.; Canada, J.M.; Buckley, L.F.; Trankle, C.R.; Billingsley, H.E.; Dixon, D.L.; Mauro, A.G.; Dessie, S.; Kadariya, D.; Mezzaroma, E.; et al. Dietary Fat, Sugar Consumption, and Cardiorespiratory Fitness in Patients with Heart Failure with Preserved Ejection Fraction. JACC Basic Transl. Sci. 2017, 2, 513–525. [Google Scholar] [CrossRef]
- Carbone, S.; Billingsley, H.E.; Canada, J.M.; Kadariya, D.; Medina de Chazal, H.; Rotelli, B.; Potere, N.; Paudel, B.; Markley, R. Unsaturated Fatty Acids to Improve Cardiorespiratory Fitness in Patients with Obesity and HFpEF: The UFA-Preserved Pilot Study. JACC Basic Transl. Sci. 2019, 4, 563–565. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Yokoyama, M.; Saito, Y.; Origasa, H.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; Kita, T.; et al. Incremental Effects of Eicosapentaenoic Acid on Cardiovascular Events in Statin-Treated Patients with Coronary Artery Disease. Circ. J. 2009, 73, 1283–1290. [Google Scholar] [CrossRef] [Green Version]
- Rizos, E.C.; Ntzani, E.E.; Bika, E.; Kostapanos, M.S.; Elisaf, M.S. Association Between Omega-3 Fatty Acid Supplementation and Risk of Major Cardiovascular Disease Events: A Systematic Review and Meta-Analysis. JAMA 2012, 308, 1024–1033. [Google Scholar] [CrossRef]
- Marchioli, R.; Barzi, F.; Bomba, E.; Chieffo, C.; Di Gregorio, D.; Di Mascio, R.; Franzosi, M.G.; Geraci, E.; Levantesi, G.; Maggioni, A.P.; et al. Early Protection Against Sudden Death by N-3 Polyunsaturated Fatty Acids After Myocardial Infarction: Time-Course Analysis of the Results of the Gruppo Italiano Per Lo Studio Della Sopravvivenza Nell’infarto Miocardico (Gissi)-Prevenzione. Circulation 2002, 105, 1897–1903. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Sakata, Y.; Nakatani, D.; Suna, S.; Usami, M.; Matsumoto, S.; Hamasaki, T.; Doi, Y.; Nishino, M.; Sato, H.; et al. Low Levels of Serum N-3 Polyunsaturated Fatty Acids Are Associated with Worse Heart Failure-Free Survival in Patients After Acute Myocardial Infarction. Circ. J. 2013, 77, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Yagi, S.; Aihara, K.; Fukuda, D.; Takashima, A.; Hara, T.; Hotchi, J.; Ise, T.; Yamaguchi, K.; Tobiume, T.; Iwase, T.; et al. Effects of Docosahexaenoic Acid on the Endothelial Function in Patients with Coronary Artery Disease. J. Atheroscler. Thromb. 2015, 22, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, J.; Sano, M.; Isobe, Y.; Fukuda, K.; Kang, X.J.; Arai, H.; Arita, M. 18-HEPE, An N-3 Fatty Acid Metabolite Released by Macrophages, Prevents Pressure Overload-Induced Maladaptive Cardiac Remodeling. J. Exp. Med. 2014, 211, 1673–1687. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C., Jr.; et al. Mitochondrial Protein Hyperacetylation in the Failing Heart. JCI Insight 2016, 2, e84897. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wang, D.D.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD Level Suppresses Inflammatory Activation of Pbmcs in Heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef]
- Abdellatif, M.; Trummer-Herbst, V.; Koser, F.; Durand, S.; Adão, R.; Vasques-Nóvoa, F.; Freundt, J.K.; Voglhuber, J.; Pricolo, M.R. Nicotinamide for The Treatment of Heart Failure with Preserved Ejection Fraction. Sci. Transl. Med. 2021, 13, eabd7064. [Google Scholar] [CrossRef]
- Oka, S.I.; Byun, J.; Huang, C.Y.; Imai, N.; Ralda, G.; Zhai, P.; Xu, X.; Kashyap, S.; Warren, J.S.; Alan Maschek, J.; et al. Nampt Potentiates Antioxidant Defense in Diabetic Cardiomyopathy. Circ. Res. 2021, 29, 114–130. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Altamirano, F.; Szweda, P.A.; Elnwasany, A.; Lee, D.I.; Yoo, H.; Kass, D.A.; Szweda, L.I.; et al. NAD+ Repletion Reverses Heart Failure with Preserved Ejection Fraction. Circ. Res. 2021, 128, 1629–1641. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, T.; Sano, M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. Int. J. Mol. Sci. 2022, 23, 996. https://doi.org/10.3390/ijms23020996
Yamamoto T, Sano M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. International Journal of Molecular Sciences. 2022; 23(2):996. https://doi.org/10.3390/ijms23020996
Chicago/Turabian StyleYamamoto, Tsunehisa, and Motoaki Sano. 2022. "Deranged Myocardial Fatty Acid Metabolism in Heart Failure" International Journal of Molecular Sciences 23, no. 2: 996. https://doi.org/10.3390/ijms23020996
APA StyleYamamoto, T., & Sano, M. (2022). Deranged Myocardial Fatty Acid Metabolism in Heart Failure. International Journal of Molecular Sciences, 23(2), 996. https://doi.org/10.3390/ijms23020996