
Diclofenac Sensitizes Signet Ring Cell Gastric Carcinoma Cells to Cisplatin by Activating Autophagy and Inhibition of Survival Signal Pathways

 and
and
Abstract
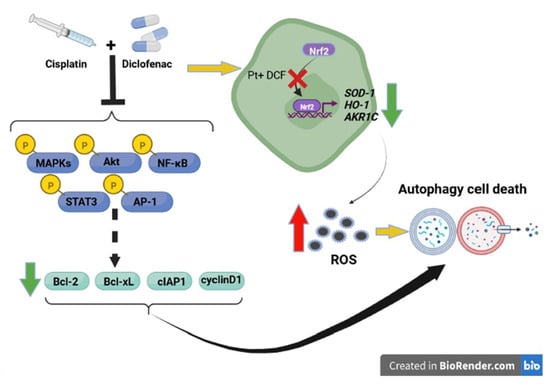
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. DCF Potentiated the Cytotoxicity of Cisplatin in Cisplatin-Resistant Signet Ring Cell Gastric Carcinoma
2.2. DCF Enhanced the Cytotoxicity of Cisplatin in KATO/DDP Cells via Autophagy Cell Death
2.3. DCF Potentiated Cytotoxicity of Cisplatin by Regenerating Intracellular ROS
2.4. DCF Downregulated Nrf2 Downstream Target Genes
2.5. DCF Potentiated Cisplatin-Induced Cytotoxicity in KATO/DDP Cells by Down Regulation of the Cell Survival Signaling Pathway
3. Discussion
4. Material and Method
4.1. Chemical and Reagents
4.2. Cell Culture Condition
4.3. Cytotoxicity Assay by Using Trypan Cell Exclusion
4.4. Apoptosis Assay
4.5. Monodensyl Cadaverine Staining
4.6. 2′,7′-Dichlorofluorescin Diacetate Assay
4.7. Western Blotting Analysis
4.8. Aldose Reductase Inhibitor Screening Assay
4.9. Bioinformatics Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DCF | Diclofenac |
| SRCGC | Signet ring cell gastric carcinoma cells |
| ROS | Reactive oxygen species |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| SOD | Superoxide dismutase |
| AKR | Aldoketoreductases |
| HO-1 | Heme oxygenase-1 |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | Fetal bovine serum |
| 3MA | 3-Methyladenine |
| MDC | Mono densylcadaverine |
| PBS | Phosphate-buffered saline |
| DMSO | Dimethyl sulfoxide |
| DCF-DA | 2′,7′-Dichlorofluorescin diacetate |
| TCGA | The Cancer Genomic Atlas |
| NAC | N-acetyl cysteine |
| NSAID | Non-steroidal anti-inflammatory drugs |
References
- Benesch, M.G.K.; Mathieson, A. Epidemiology of signet ring cell adenocarcinomas. Cancers 2020, 12, 1544. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.T.; Chen, X.Z.; Chen, Y.; Zhou, Y.W.; Tang, L.S.; Luo, D.Y.; Li, Q.; Qiu, M.; Wang, X.; Cao, D.; et al. Chemoradiotherapy is inferior to chemotherapy alone in adjuvant setting for signet ring cell containing gastric cancer. Front. Oncol. 2020, 10, 570268. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chang, J.Y. New insights into mechanisms of cisplatin resistance: From tumor cell to microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Fantone, S.; Montanari, E.; Marzioni, D.; Goteri, G. Role of Nrf2 in ovarian cancer. Antioxidants 2022, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Phoo, N.L.L.; Dejkriengkraikul, P.; Khaw-On, P.; Yodkeeree, S. Transcriptomic profiling reveals AKR1C1 and AKR1C3 mediate cisplatin resistance in signet ring cell gastric carcinoma via autophagic cell death. Int. J. Mol. Sci. 2021, 22, 12512. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Albano, F.; Arcucci, A.; Granato, G.; Romano, S.; Montagnani, S.; De Vendittis, E.; Ruocco, M.R. Markers of mitochondrial dysfunction during the diclofenac-induced apoptosis in melanoma cell lines. Biochimie 2013, 95, 934–945. [Google Scholar] [CrossRef]
- Jung, S.H.; Lee, W.; Park, S.H.; Lee, K.Y.; Choi, Y.J.; Choi, S.; Kang, D.; Kim, S.; Chang, T.S.; Hong, S.S.; et al. Diclofenac impairs autophagic flux via oxidative stress and lysosomal dysfunction: Implications for hepatotoxicity. Redox Biol. 2020, 37, 101751. [Google Scholar] [CrossRef]
- Choi, S.; Kim, S.; Park, J.; Lee, S.E.; Kim, C.; Kang, D. Diclofenac: A nonsteroidal anti-inflammatory drug inducing cancer cell death by inhibiting microtubule polymerization and autophagy flux. Antioxidants 2022, 11, 1009. [Google Scholar] [CrossRef]
- Okamoto, K.; Ueda, H.; Saito, Y.; Narumi, K.; Furugen, A.; Kobayashi, M. Diclofenac potentiates the antitumor effect of cisplatin in a xenograft mouse model transplanted with cisplatin-resistant cells without enhancing cisplatin-induced nephrotoxicity. Drug Metab. Pharmacokinet 2021, 41, 100417. [Google Scholar] [CrossRef] [PubMed]
- Gobec, S.; Brozic, P.; Rizner, T.L. Nonsteroidal anti-inflammatory drugs and their analogues as inhibitors of aldo-keto reductase AKR1C3: New lead compounds for the development of anticancer agents. Bioorg. Med. Chem. Lett. 2005, 15, 5170–5175. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.J. Diclofenac: An update on its mechanism of action and safety profile. Curr. Med. Res. Opin. 2010, 26, 1715–1731. [Google Scholar] [CrossRef] [PubMed]
- Hilovska, L.; Jendzelovsky, R.; Fedorocko, P. Potency of non-steroidal anti-inflammatory drugs in chemotherapy. Mol. Clin. Oncol. 2015, 3, 3–12. [Google Scholar] [CrossRef] [Green Version]
- El-Sheikh, A.; Khired, Z. Interactions of analgesics with cisplatin: Modulation of anticancer efficacy and potential organ toxicity. Medicina 2021, 58, 46. [Google Scholar] [CrossRef]
- Okamoto, K.; Saito, Y.; Narumi, K.; Furugen, A.; Iseki, K.; Kobayashi, M. Anticancer effects of non-steroidal anti-inflammatory drugs against cancer cells and cancer stem cells. Toxicol. In Vitro 2021, 74, 105155. [Google Scholar] [CrossRef]
- Magnano, S.; Hannon Barroeta, P.; Duffy, R.; O’Sullivan, J.; Zisterer, D.M. Cisplatin induces autophagy-associated apoptosis in human oral squamous cell carcinoma (OSCC) mediated in part through reactive oxygen species. Toxicol. Appl. Pharmacol. 2021, 427, 115646. [Google Scholar] [CrossRef]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [Green Version]
- Tanida, S.; Mizoshita, T.; Ozeki, K.; Tsukamoto, H.; Kamiya, T.; Kataoka, H.; Sakamuro, D.; Joh, T. Mechanisms of cisplatin-induced apoptosis and of cisplatin sensitivity: Potential of bin1 to act as a potent predictor of cisplatin sensitivity in gastric Cancer Treatment. Int. J. Surg. Oncol. 2012, 2012, 862879. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Tan, T.; Liu, P. Regulation of autophagy by non-steroidal anti-inflammatory drugs in cancer. Cancer Manag. Res. 2020, 12, 4595–4604. [Google Scholar] [CrossRef]
- Shen, M.; Duan, W.M.; Wu, M.Y.; Wang, W.J.; Liu, L.; Xu, M.D.; Zhu, J.; Li, D.M.; Gui, Q.; Lian, L.; et al. Participation of autophagy in the cytotoxicity against breast cancer cells by cisplatin. Oncol. Rep. 2015, 34, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Xipell, E.; Gonzalez-Huarriz, M.; Martinez de Irujo, J.J.; Garcia-Garzon, A.; Lang, F.F.; Jiang, H.; Fueyo, J.; Gomez-Manzano, C.; Alonso, M.M. Salinomycin induced ROS results in abortive autophagy and leads to regulated necrosis in glioblastoma. Oncotarget 2016, 7, 30626–30641. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Mohammadi, A.T.; Gholami, M.H.; Hashemi, F.; Zarrabi, A.; Zabolian, A.; Hushmandi, K.; Makvandi, P.; Samec, M.; Liskova, A.; et al. Nrf2 signaling pathway in cisplatin chemotherapy: Potential involvement in organ protection and chemoresistance. Pharmacol. Res. 2021, 167, 105575. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Ghosh, P.; Singha Roy, S.; Basu, A.; Bhattacharjee, A.; Bhattacharya, S. Sensitization of cisplatin therapy by a naphthalimide based organoselenium compound through modulation of antioxidant enzymes and p53 mediated apoptosis. Free Radic. Res. 2015, 49, 453–471. [Google Scholar] [CrossRef]
- Tian, H.; Li, X.; Jiang, W.; Lv, C.; Sun, W.; Huang, C.; Chen, R. High expression of AKR1C1 is associated with proliferation and migration of small-cell lung cancer cells. Lung Cancer 2016, 7, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.F.; Wang, S.G.; Zhao, Z.Y.; Li, W.L. AKR1C1-3, notably AKR1C3, are distinct biomarkers for liver cancer diagnosis and prognosis: Database mining in malignancies. Oncol. Lett. 2019, 18, 4515–4522. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.J.; Wang, J.; Zhou, J.Y.; Wu, G.S. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010, 394, 600–605. [Google Scholar] [CrossRef]
- Achkar, I.W.; Abdulrahman, N.; Al-Sulaiti, H.; Joseph, J.M.; Uddin, S.; Mraiche, F. Cisplatin based therapy: The role of the mitogen activated protein kinase signaling pathway. J. Transl. Med. 2018, 16, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Igea, A.; Canovas, B.; Dolado, I.; Nebreda, A.R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol. Med. 2013, 5, 1759–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohr, K.; Hamacher, A.; Engelke, L.H.; Kassack, M.U. Inhibition of PI3K/Akt/mTOR overcomes cisplatin resistance in the triple negative breast cancer cell line HCC38. BMC Cancer 2017, 17, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnberg, T.; Lasithiotakis, K.; Niessner, H.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Campos, M.; Gogel, J.; Garbe, C.; et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J. Investig. Dermatol. 2009, 129, 1500–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, C.; Tan, T.; Li, S.; Tang, S.; Chen, X. Sinomenine sensitizes human gastric cancer cells to cisplatin through negative regulation of PI3K/AKT/Wnt signaling pathway. Anticancer Drugs 2019, 30, 983–990. [Google Scholar] [CrossRef]
- Zhu, H.; Shi, Y.; Jiao, X.; Yang, G.; Wang, R.; Yuan, Y. Synergistic antitumor effect of dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with cisplatin on drug-resistant non-small cell lung cancer cell. Oncol. Lett. 2020, 20, 326. [Google Scholar] [CrossRef]
- Inoue, A.; Muranaka, S.; Fujita, H.; Kanno, T.; Tamai, H.; Utsumi, K. Molecular mechanism of diclofenac-induced apoptosis of promyelocytic leukemia: Dependency on reactive oxygen species, Akt, Bid, cytochrome and caspase pathway. Free Radic. Biol. Med. 2004, 37, 1290–1299. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, H.; Wang, Z.L.; Li, W.H.; Liu, H.; Zhao, Y.X. The PI3K/AKT/mTOR pathway regulates autophagy to induce apoptosis of alveolar epithelial cells in chronic obstructive pulmonary disease caused by PM2.5 particulate matter. J. Int. Med. Res. 2020, 48, 300060520927919. [Google Scholar] [CrossRef]
- Alam, M.; Mishra, R. Bcl-xL expression and regulation in the progression, recurrence, and cisplatin resistance of oral cancer. Life Sci. 2021, 280, 119705. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, L.; Lovly, C.M.; Ratner, L. The role of NF-{kappa}B-1 and NF-{kappa}B-2-mediated resistance to apoptosis in lymphomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9220–9225. [Google Scholar] [CrossRef]
- Singh, V.; Gupta, D.; Arora, R. NF-kB as a key player in regulation of cellular radiation responses and identification of radiation countermeasures. Discoveries 2015, 3, e35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Yan, Y.; Chen, M.; Luo, G.; Hao, J.; Pan, J.; Hu, S.; Guo, P.; Li, W.; Wang, R.; et al. Aspirin enhances the sensitivity of colon cancer cells to cisplatin by abrogating the binding of NF-kappaB to the COX-2 promoter. Aging 2020, 12, 611–627. [Google Scholar] [CrossRef]
- Li, Y.; He, L.R.; Gao, Y.; Zhou, N.N.; Liu, Y.; Zhou, X.K.; Liu, J.F.; Guan, X.Y.; Ma, N.F.; Xie, D. CHD1L contributes to cisplatin resistance by upregulating the ABCB1-NF-kappaB axis in human non-small-cell lung cancer. Cell Death Dis. 2019, 10, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ara, T.; Nakata, R.; Sheard, M.A.; Shimada, H.; Buettner, R.; Groshen, S.G.; Ji, L.; Yu, H.; Jove, R.; Seeger, R.C.; et al. Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer Res. 2013, 73, 3852–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limpakan Yamada, S.; Wongsirisin, P.; Yodkeeree, S.; Chakrabandhu, B.; Chongruksut, W.; Limtrakul Dejkriengkraikul, P. Interleukin-8 associated with chemosensitivity and poor chemotherapeutic response to gastric cancer. J. Gastrointest. Oncol. 2019, 10, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lae Lae Phoo, N.; Sukhamwang, A.; Dejkriengkraikul, P.; Yodkeeree, S. Diclofenac Sensitizes Signet Ring Cell Gastric Carcinoma Cells to Cisplatin by Activating Autophagy and Inhibition of Survival Signal Pathways. Int. J. Mol. Sci. 2022, 23, 12066. https://doi.org/10.3390/ijms232012066
Lae Lae Phoo N, Sukhamwang A, Dejkriengkraikul P, Yodkeeree S. Diclofenac Sensitizes Signet Ring Cell Gastric Carcinoma Cells to Cisplatin by Activating Autophagy and Inhibition of Survival Signal Pathways. International Journal of Molecular Sciences. 2022; 23(20):12066. https://doi.org/10.3390/ijms232012066
Chicago/Turabian StyleLae Lae Phoo, Nang, Amonnat Sukhamwang, Pornngarm Dejkriengkraikul, and Supachai Yodkeeree. 2022. "Diclofenac Sensitizes Signet Ring Cell Gastric Carcinoma Cells to Cisplatin by Activating Autophagy and Inhibition of Survival Signal Pathways" International Journal of Molecular Sciences 23, no. 20: 12066. https://doi.org/10.3390/ijms232012066
APA StyleLae Lae Phoo, N., Sukhamwang, A., Dejkriengkraikul, P., & Yodkeeree, S. (2022). Diclofenac Sensitizes Signet Ring Cell Gastric Carcinoma Cells to Cisplatin by Activating Autophagy and Inhibition of Survival Signal Pathways. International Journal of Molecular Sciences, 23(20), 12066. https://doi.org/10.3390/ijms232012066