
E2F4DN Transgenic Mice: A Tool for the Evaluation of E2F4 as a Therapeutic Target in Neuropathology and Brain Aging

 , , ,
, , , 
Abstract
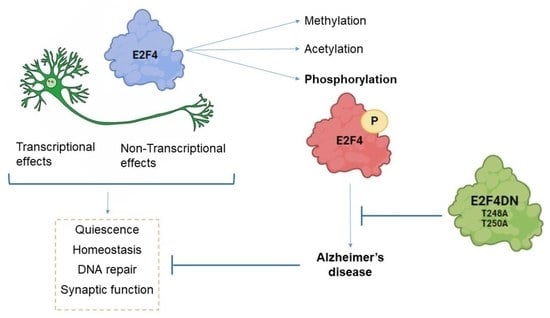
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Transcriptional and Non-Transcriptional Functions of E2F4
2.1. E2F4 as a Transcriptional Regulator
2.2. E2F4 and Non-Transcriptional Interactors
3. Regulation of E2F4 Function by Chemical Modifications
4. E2F4 as a Multifactorial Regulator
4.1. E2F4 as a Regulator of Tissue Homeostasis
4.2. E2F4 as a Regulator of DNA Repair
4.3. E2F4 as a Putative Regulator of Synaptic Function
4.3.1. Transcriptional Regulation of Synaptic Function by E2F4
4.3.2. Interaction of E2F4 with Synaptic Regulators
4.3.3. E2F4 and MAPK Proteins in Synaptic Function
5. E2F4 and Alzheimer’s Disease (AD)
5.1. AD Etiology
5.2. Connection of E2F4 with AD
5.2.1. E2F4, Cell Cycle Re-Entry, and Neuronal Tetraploidy
5.2.2. E2F4 and Neuroinflammation, Aβ Peptide Proteostasis, and Body Weight Loss
5.2.3. E2F4 and Cognitive Impairment
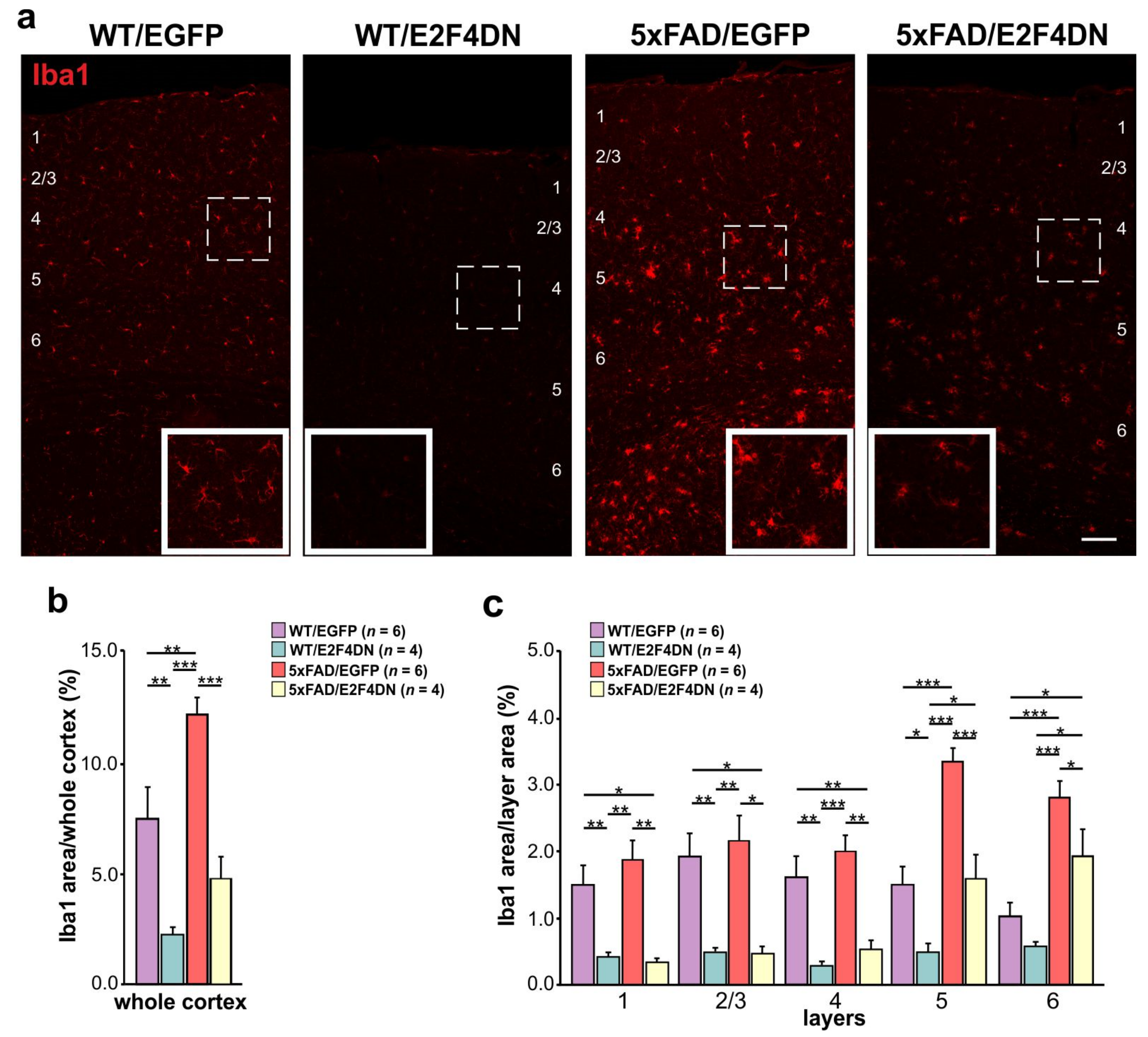
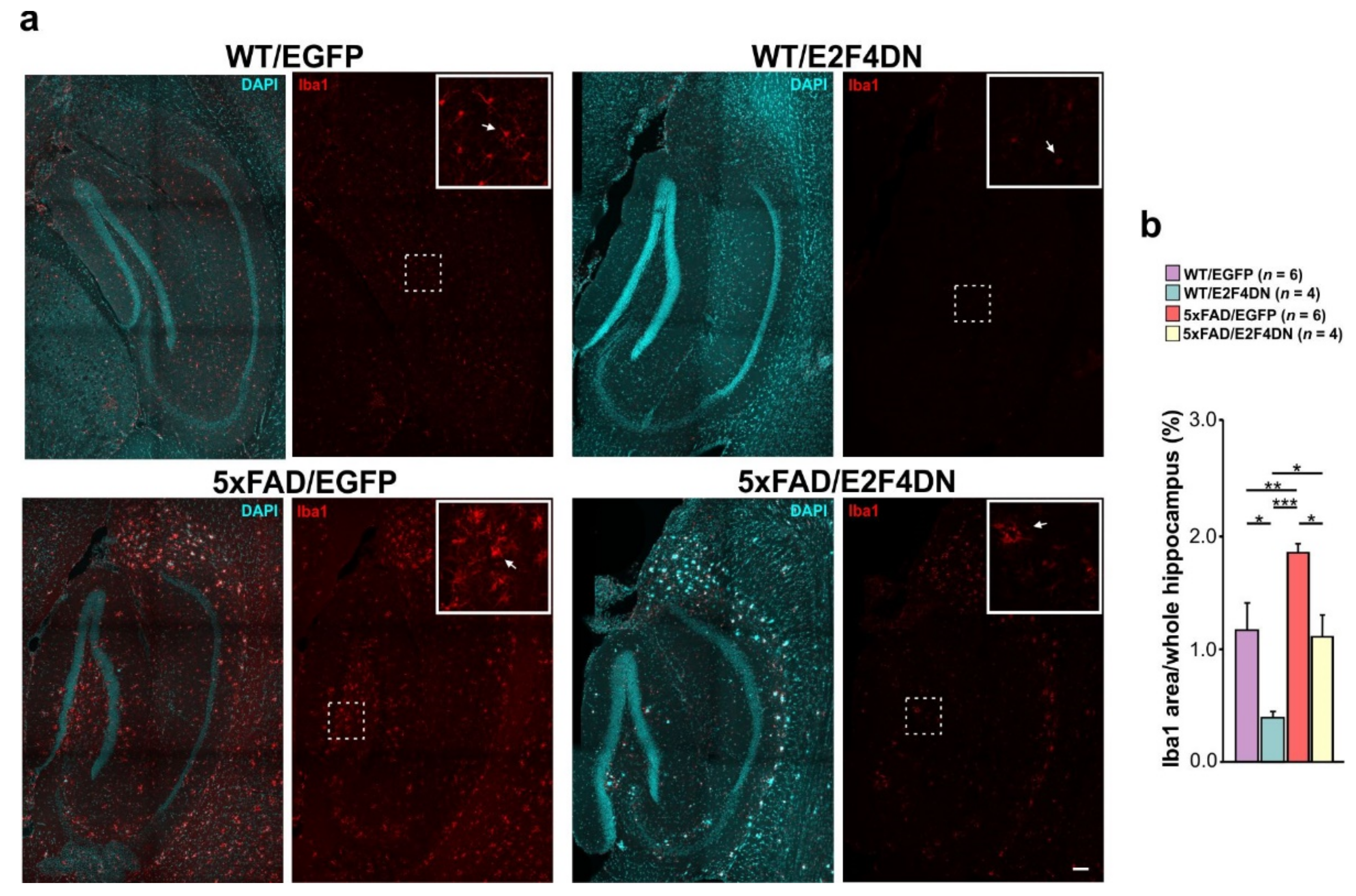
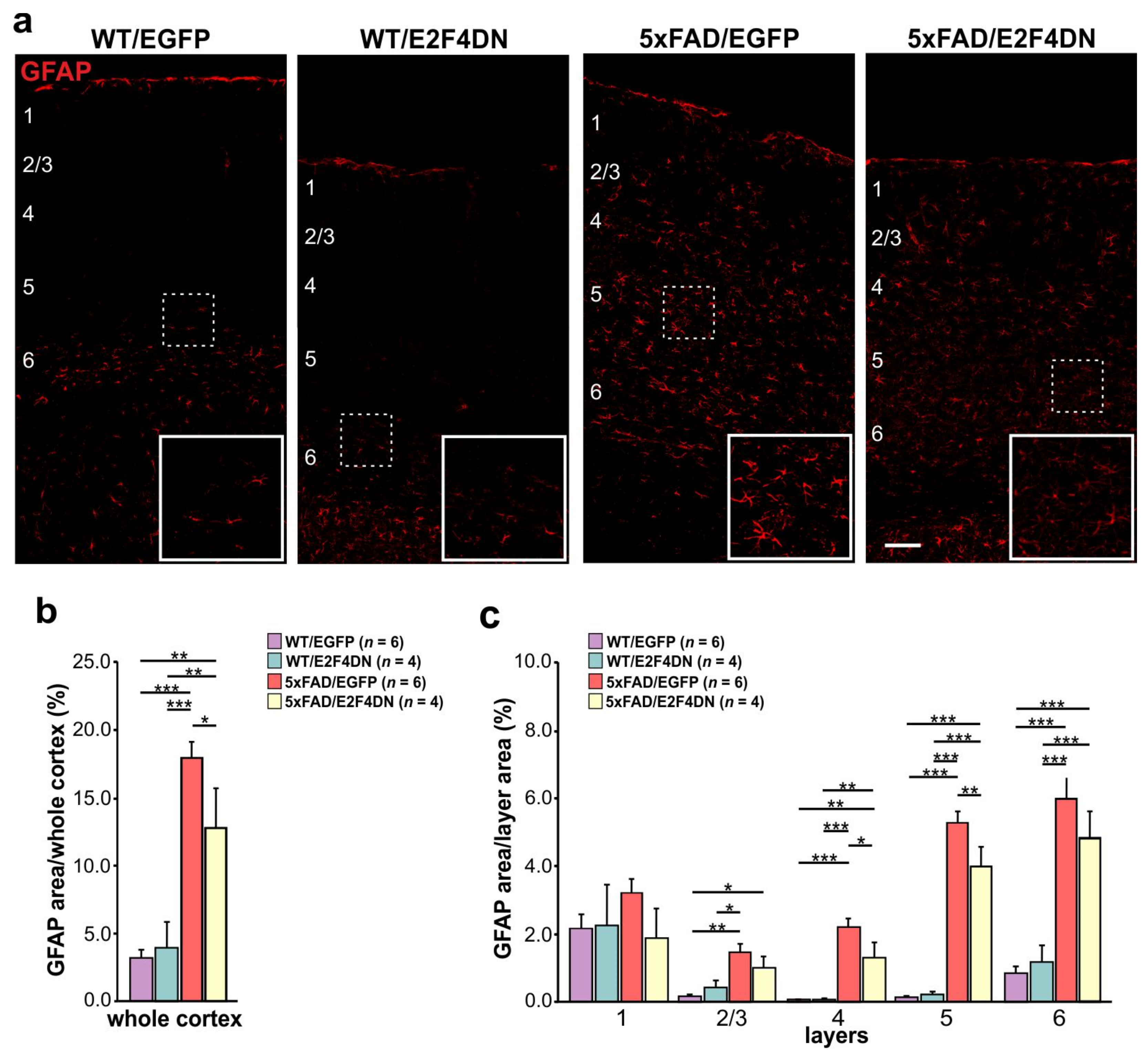
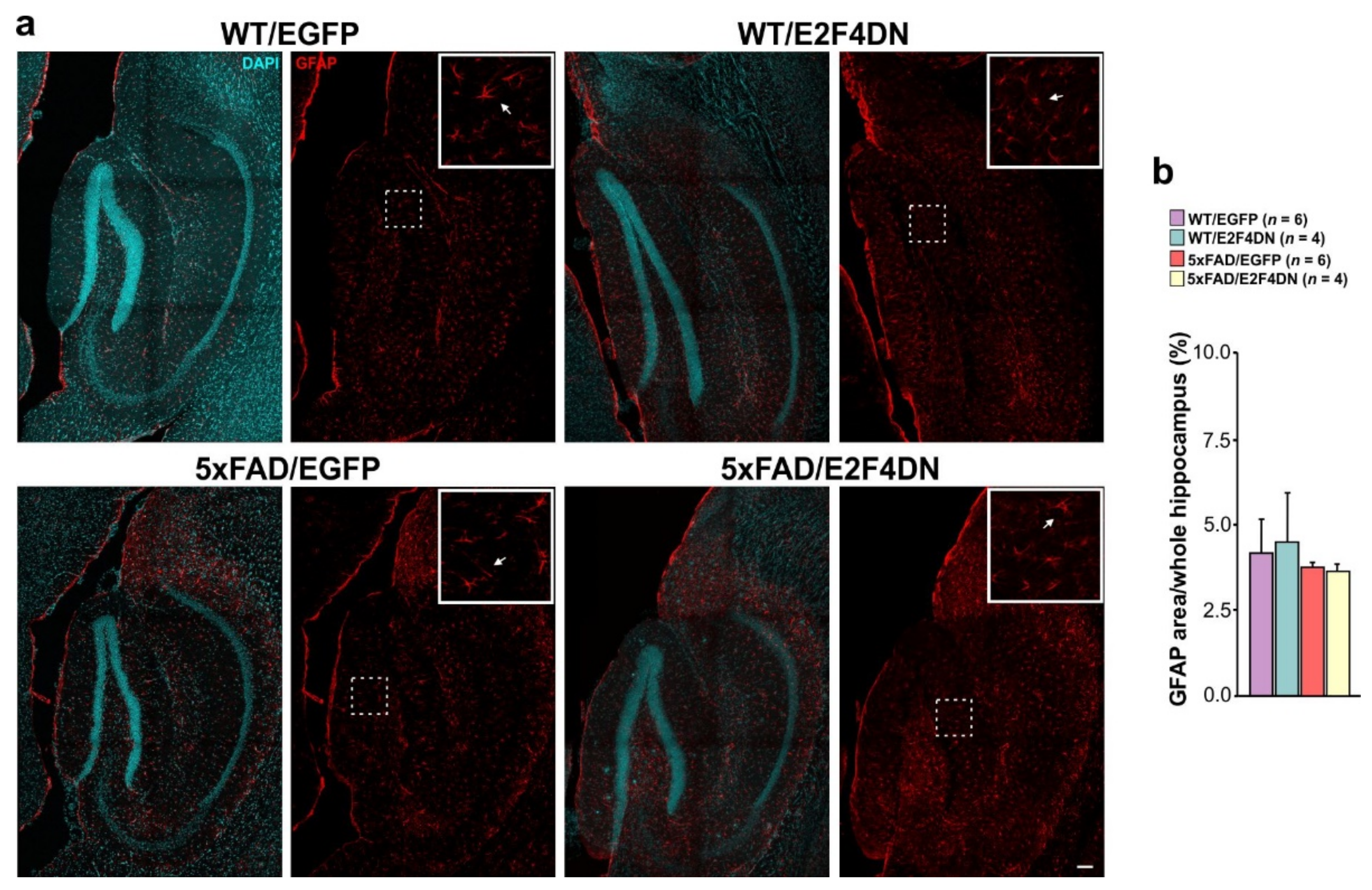
6. E2F4DN Transgenic Mice and Neuroinflammation
6.1. E2F4DN and Microgliosis
6.2. E2F4DN and Reactive Astrogliosis
7. Discussion
8. Materials and Methods
8.1. Mice
8.2. Antibodies
8.3. Tissue Processing
8.4. Immunohistochemistry
8.5. Quenching of Lipofuscin Autofluorescence Signal
8.6. Confocal Microscopy and Image Analysis
8.7. Statistical Analysis
8.8. Gene Ontology Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierce, A.M.; Schneider-Broussard, R.; Philhower, J.L.; Johnson, D.G. Differential activities of E2F family members: Unique functions in regulating transcription. Mol. Carcinog. 1998, 22, 190–198. [Google Scholar] [CrossRef]
- Ginsberg, D.; Vairo, G.; Chittenden, T.; Xiao, Z.X.; Xu, G.; Wydner, K.L.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 1994, 8, 2665–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beijersbergen, R.L.; Kerkhoven, R.M.; Zhu, L.; Carlée, L.; Voorhoeve, P.M.; Bernards, R. E2F-4, a new member of the E2F gene family, has oncogenic activity and associates with p107 in vivo. Genes Dev. 1994, 8, 2680–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vairo, G.; Livingston, D.M.; Ginsberg, D. Functional interaction between E2F-4 and p130: Evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 1995, 9, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Garneau, H.; Paquin, M.C.; Carrier, J.C.; Rivard, N. E2F4 expression is required for cell cycle progression of normal intestinal crypt cells and colorectal cancer cells. J. Cell Physiol. 2009, 221, 350–358. [Google Scholar] [CrossRef]
- van Amerongen, M.J.; Diehl, F.; Novoyatleva, T.; Patra, C.; Engel, F.B. E2F4 is required for cardiomyocyte proliferation. Cardiovasc. Res. 2010, 86, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Crosby, M.E.; Almasan, A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol. Ther. 2004, 3, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Sage, J. Novel functions for the transcription factor E2F4 in development and disease. Cell Cycle 2016, 15, 3183–3190. [Google Scholar] [CrossRef] [Green Version]
- Miles, S.; Breeden, L. A common strategy for initiating the transition from proliferation to quiescence. Curr. Genet. 2017, 63, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Stevaux, O.; Dyson, N.J. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 2002, 14, 684–691. [Google Scholar] [CrossRef]
- López-Sánchez, N.; Garrido-García, A.; Ramón-Landreau, M.; Cano-Daganzo, V.; Frade, J.M. E2F4-based gene therapy mitigates the phenotype of the Alzheimer’s disease mouse model 5xFAD. Neurotherapeutics 2021, 18, 2484–2503. [Google Scholar] [CrossRef]
- López-Sánchez, N.; Ramón-Landreau, M.; Trujillo, C.; Garrido-García, A.; Frade, J.M. A mutant variant of E2F4 triggers multifactorial therapeutic effects in 5xFAD mice. Mol. Neurobiol. 2022, 59, 3016–3039. [Google Scholar] [CrossRef]
- Morillo, S.M.; Abanto, E.P.; Román, M.J.; Frade, J.M. Nerve growth factor-induced cell cycle reentry in newborn neurons is triggered by p38MAPK-dependent E2F4 phosphorylation. Mol. Cell. Biol. 2012, 32, 2722–2737. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [Green Version]
- Hagan, C. When Are Mice Considered Old? 2017. Available online: https://www.jax.org/news-and-insights/jax-blog/2017/november/when-are-mice-considered-old (accessed on 28 August 2022).
- Lang, S.E.; McMahon, S.B.; Cole, M.D.; Hearing, P. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J. Biol. Chem. 2001, 276, 32627–32634. [Google Scholar] [CrossRef] [Green Version]
- Verona, R.; Moberg, K.; Estes, S.; Starz, M.; Vernon, J.P.; Lees, J.A. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol. Cell. Biol. 1997, 17, 7268–7282. [Google Scholar] [CrossRef] [Green Version]
- Gaubatz, S.; Lees, J.A.; Lindeman, G.J.; Livingston, D.M. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol. Cell. Biol. 2001, 21, 1384–1392. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Arand, J.; Chaikovsky, A.; Mooney, N.A.; Demeter, J.; Brison, C.M.; Oliverio, R.; Vogel, H.; Rubin, S.M.; Jackson, P.K.; et al. E2F4 regulates transcriptional activation in mouse embryonic stem cells independently of the RB family. Nat. Commun. 2019, 10, 2939. [Google Scholar] [CrossRef] [Green Version]
- Chook, Y.M.; Blobel, G. Karyopherins and nuclear import. Curr. Opin. Struct. Biol. 2001, 11, 703–715. [Google Scholar] [CrossRef]
- Avis, J.M.; Clarke, P.R. Ran, a GTPase involved in nuclear processes: Its regulators and effectors. J. Cell Sci. 1996, 109 Pt 10, 2423–2427. [Google Scholar] [CrossRef]
- Puri, P.L.; Cimino, L.; Fulco, M.; Zimmerman, C.; La Thangue, N.B.; Giordano, A.; Graessmann, A.; Levrero, M. Regulation of E2F4 mitogenic activity during terminal differentiation by its heterodimerization partners for nuclear translocation. Cancer Res. 1998, 58, 1325–1331. [Google Scholar]
- Lindeman, G.J.; Gaubatz, S.; Livingston, D.M.; Ginsberg, D. The subcellular localization of E2F-4 is cell-cycle dependent. Proc. Natl. Acad. Sci. USA 1997, 94, 5095–5100. [Google Scholar] [CrossRef] [Green Version]
- Magae, J.; Wu, C.L.; Illenye, S.; Harlow, E.; Heintz, N.H. Nuclear localization of DP and E2F transcription factors by heterodimeric partners and retinoblastoma protein family members. J. Cell Sci. 1996, 109, 1717–1726. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Tomita, M.; Yanagawa, H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shutling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA 2009, 106, 10171–10176. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, M.D.; Ivanova, I.A.; Dagnino, C.; D’Souza, S.J.; Dagnino, L. Active nuclear import and export pathways regulate E2F-5 subcellular localization. J. Biol. Chem. 2002, 277, 34471–34479. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.K.; Bhinge, A.A.; Iyer, V.R. Wide-ranging functions of E2F4 in transcriptional activation and repression revealed by genome-wide analysis. Nucl. Acids Res. 2011, 39, 3558–3573. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Bieda, M.; Jin, V.X.; Rabinovich, A.; Oberley, M.J.; Green, R.; Farnham, P.J. A comprehensive ChIP-chip analysis of E2F1; E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2007, 17, 1550–1561. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julian, L.M.; Liu, Y.; Pakenham, C.A.; Dugal-Tessier, D.; Ruzhynsky, V.; Bae, S.; Tsai, S.Y.; Leone, G.; Slack, R.S.; Blais, A. Tissue-specific targeting of cell fate regulatory genes by E2f factors. Cell Death Differ. 2016, 23, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; González, Y.R.; Qu, D.; Huang, E.; Safarpour, F.; Wang, E.; Joselin, A.; Im, D.S.; Callaghan, S.M.; Boonying, W.; et al. The pro-death role of Cited2 in stroke is regulated by E2F1/4 transcription factors. J. Biol. Chem. 2019, 294, 8617–8629. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, A.; Jin, V.X.; Rabinovich, R.; Xu, X.; Farnham, P.J. E2F in vivo binding specificity: Comparison of consensus versus nonconsensus binding sites. Genome Res. 2008, 18, 1763–1777. [Google Scholar] [CrossRef]
- Tommasi, S.; Pfeifer, G.P. In vivo structure of the human cdc2 promoter: Release of a p130-E2F-4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of cdc2 expression. Mol. Cell. Biol. 1995, 15, 6901–6913. [Google Scholar] [CrossRef] [Green Version]
- Tuncay, K.; Ensman, L.; Sun, J.; Haidar, A.A.; Stanley, F.; Trelinski, M.; Ortoleva, P. Transcriptional regulatory networks via gene ontology and expression data. In Silico Biol. 2007, 7, 21–34. [Google Scholar]
- Feng, Y.; Li, L.; Du, Y.; Peng, X.; Chen, F. E2F4 functions as a tumour suppressor in acute myeloid leukaemia via inhibition of the MAPK signalling pathway by binding to EZH2. J. Cell. Mol. Med. 2020, 24, 2157. [Google Scholar] [CrossRef] [Green Version]
- Gill, R.M.; Hamel, P.A. Subcellular compartmentalization of E2F family members is required for maintenance of the postmitotic state in terminally differentiated muscle. J. Cell Biol. 2000, 148, 1187–1201. [Google Scholar] [CrossRef] [Green Version]
- Hazan, R.; Mori, M.; Danielian, P.S.; Guen, V.J.; Rubin, S.M.; Cardoso, W.V.; Lees, J.A. E2F4’s cytoplasmic role in multiciliogenesis is mediated via an N-terminal domain that binds two components of the centriole replication machinery, Deup1 and SAS6. Mol. Biol. Cell 2021, 32, ar1. [Google Scholar] [CrossRef]
- Mori, M.; Hazan, R.; Danielian, P.S.; Mahoney, J.E.; Li, H.; Lu, J.; Miller, E.S.; Zhu, X.; Lees, J.A.; Cardoso, W.V. Cytoplasmic E2f4 forms organizing centres for initiation of centriole amplification during multiciliogenesis. Nat. Commun. 2017, 8, 15857. [Google Scholar] [CrossRef] [Green Version]
- Stracker, T.H. E2F4/5-mediated transcriptional control of multiciliated cell differentiation: Redundancy or fine-tuning? Dev. Biol. 2019, 446, 20–21. [Google Scholar] [CrossRef]
- Ma, L.; Quigley, I.; Omran, H.; Kintner, C. Multicilin drives centriole biogenesis via E2f proteins. Genes Dev. 2014, 28, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Chong, Y.L.; Zhang, Y.; Zhou, F.; Roy, S. Distinct requirements of E2f4 versus E2f5 activity for multiciliated cell development in the zebrafish embryo. Dev. Biol. 2018, 443, 165–172. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.A.; Wang, Y.C.; Chuang, Y.P.; Huang, Y.H.; Su, W.C.; Chang, W.C.; Hung, J.J. EGF-mediated inhibition of ubiquitin-specific peptidase 24 expression has a crucial role in tumorigenesis. Oncogene 2017, 36, 2930–2945. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Balbás, M.A.; Bauer, U.M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 activity by acetylation. EMBO J. 2000, 19, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Carr, S.M.; Poppy Roworth, A.; Chan, C.; La Thangue, N.B. Post-translational control of transcription factors: Methylation ranks highly. FEBS J. 2015, 282, 4450–4465. [Google Scholar] [CrossRef]
- Kontaki, H.; Talianidis, I. Lysine methylation regulates E2F1-induced cell death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef]
- Xie, Q.; Bai, Y.; Wu, J.; Sun, Y.; Wang, Y.; Zhang, Y.; Mei, P.; Yuan, Z. Methylation-mediated regulation of E2F1 in DNA damage-induced cell death. J. Recept. Signal Transduct. Res. 2011, 31, 139–146. [Google Scholar] [CrossRef]
- Advani, S.J.; Weichselbaum, R.R.; Roizman, B. E2F proteins are posttranslationally modified concomitantly with a reduction in nuclear binding activity in cells infected with herpes simplex virus 1. J Virol. 2000, 74, 7842–7850. [Google Scholar] [CrossRef] [Green Version]
- Scimè, A.; Li, L.; Ciavarra, G.; Whyte, P. Cyclin D1/cdk4 can interact with E2F4/DP1 and disrupts its DNA-binding capacity. J. Cell Physiol. 2008, 214, 568–581. [Google Scholar] [CrossRef]
- Araki, K.; Kawauchi, K.; Tanaka, N. IKK/NF-kappaB signaling pathway inhibits cell-cycle progression by a novel Rb-independent suppression system for E2F transcription factors. Oncogene 2008, 27, 5696–5705. [Google Scholar] [CrossRef] [Green Version]
- Paquin, M.C.; Cagnol, S.; Carrier, J.C.; Leblanc, C.; Rivard, N. ERK-associated changes in E2F4 phosphorylation, localization and transcriptional activity during mitogenic stimulation in human intestinal epithelial crypt cells. BMC Cell Biol. 2013, 14, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Brauer, C.; Chen, Y.J.; Brauer, P.M.; Pimkina, J.; Raychaudhuri, P. ARF stimulates XPC to trigger nucleotide excision repair by regulating the repressor complex of E2F4. EMBO Rep. 2009, 10, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11, eaav4508. [Google Scholar] [CrossRef] [PubMed]
- Dingar, D.; Konecny, F.; Zou, J.; Sun, X.; von Harsdorf, R. Anti-apoptotic function of the E2F transcription factor 4 (E2F4)/p130, a member of retinoblastoma gene family in cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 53, 820–828. [Google Scholar] [CrossRef]
- Luo, C.; Sheng, J.; Hu, M.G.; Haluska, F.G.; Cui, R.; Xu, Z.; Tsichlis, P.N.; Hu, G.F.; Hinds, P.W. Loss of ARF sensitizes transgenic BRAFV600E mice to UV-induced melanoma via suppression of XPC. Cancer Res. 2013, 73, 4337–4348. [Google Scholar] [CrossRef] [Green Version]
- Pamuklar, Z.N.; Chen, J.; Muehlbauer, M.; Spagnoli, A.; Torquati, A. Necdin-E2F4 interaction provides insulin-sensitizing effect after weight loss induced by gastric bypass surgery. Surg. Obes. Relat. Dis. 2013, 9, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.D.; Zan, L.S.; Li, A.N.; Cheng, G.; Li, S.J.; Zhang, Y.R.; Wang, X.Y.; Zhang, Y.Y. Characterization of the promoter region of the bovine long-chain acyl-CoA synthetase 1 gene: Roles of E2F1, Sp1, KLF15, and E2F4. Sci. Rep. 2016, 6, 19661. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.N.; Huang, G.Y.; Zhou, J.; Mao, W.; Langley, R.; Lu, Z.; Bast, R.C., Jr. Amino Acid Deprivation-Induced Autophagy Requires Upregulation of DIRAS3 through Reduction of E2F1 and E2F4 Transcriptional Repression. Cancers 2019, 11, 603. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, O.; Moll, U.M. Macrophage migration inhibitory factor MIF interferes with the Rb-E2F pathway. Mol. Cell 2005, 17, 225–236. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Xue, M.; Tang, Y.; Xu, J.; Liu, L.; Huang, Y.; Yang, Y.; Qiu, H.; Guo, F. Overexpressing p130/E2F4 in mesenchymal stem cells facilitates the repair of injured alveolar epithelial cells in LPS-induced ARDS mice. Stem Cell Res. Ther. 2019, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Khozoie, C.; Bility, M.T.; Ferry, C.H.; Blazanin, N.; Glick, A.B.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor β/δ cross talks with E2F and attenuates mitosis in HRAS-expressing cells. Mol. Cell. Biol. 2012, 32, 2065–2082. [Google Scholar] [CrossRef] [Green Version]
- Scimè, A.; Soleimani, V.D.; Bentzinger, C.F.; Gillespie, M.A.; Le Grand, F.; Grenier, G.; Bevilacqua, L.; Harper, M.E.; Rudnicki, M.A. Oxidative status of muscle is determined by p107 regulation of PGC-1alpha. J. Cell Biol. 2010, 190, 651–662. [Google Scholar] [CrossRef]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Sweeney, G.; Song, J. The association between PGC-1α and Alzheimer’s disease. Anat. Cell Biol. 2016, 49, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Cam, H.; Balciunaite, E.; Blais, A.; Spektor, A.; Scarpulla, R.C.; Young, R.; Kluger, Y.; Dynlacht, B.D. A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell 2004, 16, 399–411. [Google Scholar] [CrossRef]
- Puri, P.L.; Balsano, C.; Burgio, V.L.; Chirillo, P.; Natoli, G.; Ricci, L.; Mattei, E.; Graessmann, A.; Levrero, M. MyoD prevents cyclinA/cdk2 containing E2F complexes formation in terminally differentiated myocytes. Oncogene 1997, 14, 1171–1184. [Google Scholar] [CrossRef] [Green Version]
- Parakati, R.; DiMario, J.X. Dynamic transcriptional regulatory complexes, including E2F4, p107, p130, and Sp1, control fibroblast growth factor receptor 1 gene expression during myogenesis. J. Biol. Chem. 2005, 280, 21284–21294. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Wang, G.; Diao, Y.; Long, Y.; Fu, X.; Weng, M.; Zhou, L.; Sun, K.; Cheung, T.H.; Ip, N.Y.; et al. A molecular switch regulating cell fate choice between muscle progenitor cells and brown adipocytes. Dev. Cell 2017, 41, e5–e391. [Google Scholar] [CrossRef] [Green Version]
- Persengiev, S.P.; Kondova, I.I.; Kilpatrick, D.L. E2F4 actively promotes the initiation and maintenance of nerve growth factor-induced cell differentiation. Mol. Cell Biol. 1999, 19, 6048–6056. [Google Scholar] [CrossRef] [Green Version]
- Fajas, L.; Landsberg, R.L.; Huss-Garcia, Y.; Sardet, C.; Lees, J.A.; Auwerx, J. E2Fs regulate adipocyte differentiation. Dev. Cell 2002, 3, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Landsberg, R.L.; Sero, J.E.; Danielian, P.S.; Yuan, T.L.; Lee, E.Y.; Lees, J.A. The role of E2F4 in adipogenesis is independent of its cell cycle regulatory activity. Proc. Natl. Acad. Sci. USA 2003, 100, 2456–2461. [Google Scholar] [CrossRef] [Green Version]
- Miard, S.; Fajas, L. Atypical transcriptional regulators and cofactors of PPARgamma. Int. J. Obes. 2005, 29 (Suppl. S1), S10–S12. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Butte, A.J.; Kokkotou, E.; Yechoor, V.K.; Taniguchi, C.M.; Kriauciunas, K.M.; Cypess, A.M.; Niinobe, M.; Yoshikawa, K.; Patti, M.E.; et al. Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and necdin. Nat. Cell Biol. 2005, 7, 601–611. [Google Scholar] [CrossRef]
- Enos, M.E.; Bancos, S.A.; Bushnell, T.; Crispe, I.N. E2F4 modulates differentiation and gene expression in hematopoietic progenitor cells during commitment to the lymphoid lineage. J. Immunol. 2008, 180, 3699–3707. [Google Scholar] [CrossRef] [Green Version]
- Yanagino, T.; Yuasa, K.; Nagahama, M.; Matsuda, Y.; Tsuji, A. Transcriptional regulation of fibrillin-2 gene by E2F family members in chondrocyte differentiation. J. Cell. Biochem. 2009, 106, 580–588. [Google Scholar] [CrossRef]
- Lee, E.Y.; Yuan, T.L.; Danielian, P.S.; West, J.C.; Lees, J.A. E2F4 cooperates with pRB in the development of extra-embryonic tissues. Dev. Biol. 2009, 332, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Hitzel, J.; Moll, F.; Kruse, C.; Malik, R.A.; Preussner, J.; Looso, M.; Leisegang, M.S.; Steinhilber, D.; Brandes, R.P.; et al. The histone demethylase PHF8 is essential for endothelial cell migration. PLoS ONE 2016, 11, e0146645. [Google Scholar] [CrossRef]
- Zhen, Z.; Zhang, M.; Yuan, X.; Li, M. Transcription factor E2F4 is a positive regulator of milk biosynthesis and proliferation of bovine mammary epithelial cells. Cell Biol. Int. 2020, 44, 229–241. [Google Scholar] [CrossRef]
- Danielian, P.S.; Bender Kim, C.F.; Caron, A.M.; Vasile, E.; Bronson, R.T.; Lees, J.A. E2f4 is required for normal development of the airway epithelium. Dev. Biol. 2007, 305, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Danielian, P.S.; Hess, R.A.; Lees, J.A. E2f4 and E2f5 are essential for the development of the male reproductive system. Cell Cycle 2016, 15, 250–260. [Google Scholar] [CrossRef] [Green Version]
- Kastner, A.; Espanel, X.; Brun, G. Transient accumulation of retinoblastoma/E2F-1 protein complexes correlates with the onset of neuronal differentiation in the developing quail neural retina. Cell Growth Differ. 1998, 9, 857–867. [Google Scholar] [PubMed]
- Francesconi, C.M.; Hutcheon, A.E.; Chung, E.H.; Dalbone, A.C.; Joyce, N.C.; Zieske, J.D. Expression patterns of retinoblastoma and E2F family proteins during corneal development. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1054–1062. [Google Scholar]
- Ruzhynsky, V.A.; McClellan, K.A.; Vanderluit, J.L.; Jeong, Y.; Furimsky, M.; Park, D.S.; Epstein, D.J.; Wallace, V.A.; Slack, R.S. Cell cycle regulator E2F4 is essential for the development of the ventral telencephalon. J. Neurosci. 2007, 27, 5926–5935. [Google Scholar] [CrossRef] [PubMed]
- Ruzhynsky, V.A.; Furimsky, M.; Park, D.S.; Wallace, V.A.; Slack, R.S. E2F4 is required for early eye patterning. Dev. Neurosci. 2009, 31, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Feng, Y.; Zhang, Y.; Feng, J. DNA damage checkpoint and repair: From the budding yeast Saccharomyces cerevisiae to the pathogenic fungus Candida albicans. Comput. Struct. Biotechnol. J. 2021, 19, 6343–6354. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef] [Green Version]
- Bukowska, B.; Mokra, K.; Michałowicz, J. Benzo[a]pyrene-Environmental Occurrence, Human Exposure, and Mechanisms of Toxicity. Int. J. Mol. Sci. 2022, 23, 6348. [Google Scholar] [CrossRef]
- Allmann, S.; Mayer, L.; Olma, J.; Kaina, B.; Hofmann, T.G.; Tomicic, M.T.; Christmann, M. Benzo[a]pyrene represses DNA repair through altered E2F1/E2F4 function marking an early event in DNA damage-induced cellular senescence. Nucl. Acids Res. 2020, 48, 12085–12101. [Google Scholar] [CrossRef]
- Shell, S.M.; Hawkins, E.K.; Tsai, M.S.; Hlaing, A.S.; Rizzo, C.J.; Chazin, W.J. Xeroderma pigmentosum complementation group C protein (XPC) serves as a general sensor of damaged DNA. DNA Repair 2013, 12, 947–953. [Google Scholar] [CrossRef] [Green Version]
- Bindra, R.S.; Glazer, P.M. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene 2007, 26, 2048–2057. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.S.; Baek, M.; Lee, D.Y.; Bang, Y.J.; Cho, H.N.; Lee, Y.S.; Ha, J.H.; Kim, H.Y.; Jeoung, D.I. MAPK signaling is involved in camptothecin-induced cell death. Mol. Cells 2002, 14, 348–354. [Google Scholar]
- Muiño, E.; Maisterra, O.; Jiménez-Balado, J.; Cullell, N.; Carrera, C.; Torres-Aguila, N.P.; Cárcel-Márquez, J.; Gallego-Fabrega, C.; Lledós, M.; González-Sánchez, J.; et al. Genome-wide transcriptome study in skin biopsies reveals an association of E2F4 with cadasil and cognitive impairment. Sci. Rep. 2021, 11, 6846. [Google Scholar] [CrossRef]
- Ding, J.; Kong, W.; Mou, X.; Wang, S. Construction of Transcriptional Regulatory Network of Alzheimer’s Disease Based on PANDA Algorithm. Interdiscip. Sci. 2019, 11, 226–236. [Google Scholar] [CrossRef]
- Iyirhiaro, G.O.; Zhang, Y.; Estey, C.; O’Hare, M.J.; Safarpour, F.; Parsanejad, M.; Wang, S.; Abdel-Messih, E.; Callaghan, S.M.; During, M.J.; et al. Regulation of ischemic neuronal death by E2F4-p130 protein complexes. J. Biol. Chem. 2014, 289, 18202–18213. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Huang, Y.; Zhang, P.; Han, C.; Lu, Y.; Mo, Z.; Zhang, Z.; Li, X.; Zhao, S.; Cai, F.; et al. Characterization of a pathogenic variant in GBA for Parkinson’s disease with mild cognitive impairment patients. Mol. Brain 2020, 13, 102. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Knafo, S.; Sánchez-Puelles, C.; Palomer, E.; Delgado, I.; Draffin, J.E.; Mingo, J.; Wahle, T.; Kaleka, K.; Mou, L.; Pereda-Perez, I.; et al. PTEN recruitment controls synaptic and cognitive function in Alzheimer’s models. Nat. Neurosci. 2016, 19, 443–453. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Tu, X.; Yasuda, R.; Colgan, L.A. Rac1 is a downstream effector of PKCα in structural synaptic plasticity. Sci. Rep. 2020, 10, 1777. [Google Scholar] [CrossRef] [Green Version]
- Lo, L.H.Y.; Dong, R.; Lyu, Q.; Lai, K.O. The Protein Arginine Methyltransferase PRMT8 and Substrate G3BP1 Control Rac1-PAK1 Signaling and Actin Cytoskeleton for Dendritic Spine Maturation. Cell Rep. 2020, 31, 107744. [Google Scholar] [CrossRef]
- O’Neil, S.D.; Rácz, B.; Brown, W.E.; Gao, Y.; Soderblom, E.J.; Yasuda, R.; Soderling, S.H. Action potential-coupled Rho GTPase signaling drives presynaptic plasticity. eLife 2021, 10, e63756. [Google Scholar] [CrossRef]
- Gauthier-Campbell, C.; Bredt, D.S.; Murphy, T.H.; El-Din El-Husseini, A. Regulation of Dendritic Branching and Filopodia Formation in Hippocampal Neurons by Specific Acylated Protein Motifs. Mol. Biol. Cell 2004, 15, 2205–2217. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Ueda, H.H.; Eto, K.; Onda, M.; Sato, A.; Ohba, T.; Nabekura, J.; Murakoshi, H. Photoactivatable CaMKII induces synaptic plasticity in single synapses. Nat. Commun. 2021, 12, 751. [Google Scholar] [CrossRef]
- Foley, K.; McKee, C.; Nairn, A.C.; Xia, H. Regulation of Synaptic Transmission and Plasticity by Protein Phosphatase 1. J. Neurosci. 2021, 41, 3040–3050. [Google Scholar] [CrossRef]
- Chiu, A.M.; Wang, J.; Fiske, M.P.; Hubalkova, P.; Barse, L.; Gray, J.A.; Sanz-Clemente, A. NMDAR-Activated PP1 Dephosphorylates GluN2B to Modulate NMDAR Synaptic Content. Cell Rep. 2019, 28, e5–e341. [Google Scholar] [CrossRef]
- Liu, X.; Kumar, V.; Tsai, N.P.; Auerbach, B.D. Hyperexcitability and Homeostasis in Fragile X Syndrome. Front. Mol. Neurosci. 2022, 14, 805929. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Bassell, G.J. Diversity on location. eLife 2022, 11, e76818. [Google Scholar] [CrossRef] [PubMed]
- Hale, C.R.; Sawicka, K.; Mora, K.; Fak, J.J.; Kang, J.J.; Cutrim, P.; Cialowicz, K.; Carroll, T.S.; Darnell, R.B. FMRP regulates mRNAs encoding distinct functions in the cell body and dendrites of CA1 pyramidal neurons. eLife 2021, 10, e71892. [Google Scholar] [CrossRef] [PubMed]
- Zalfa, F.; Eleuteri, B.; Dickson, K.S.; Mercaldo, V.; De Rubeis, S.; di Penta, A.; Tabolacci, E.; Chiurazzi, P.; Neri, G.; Grant, S.G.; et al. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci. 2007, 10, 578–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnöder, L.; Tomic, I.; Schwindt, L.; Helm, D.; Rettel, M.; Schulz-Schaeffer, W.; Krause, E.; Rettig, J.; Fassbender, K.; Liu, Y. P38α-MAPK phosphorylates Snapin and reduces Snapin-mediated BACE1 transportation in APP-transgenic mice. FASEB J. 2021, 35, e21691. [Google Scholar] [CrossRef]
- Ye, X.; Feng, T.; Tammineni, P.; Chang, Q.; Jeong, Y.Y.; Margolis, D.J.; Cai, H.; Kusnecov, A.; Cai, Q. Regulation of Synaptic Amyloid-β Generation through BACE1 Retrograde Transport in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 2639–2655. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.A.; Liu, D.X.; Troy, C.M.; Biswas, S.C. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim. Biophys. Acta 2007, 1772, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Snigdha, S.; Prieto, G.A.; Petrosyan, A.; Loertscher, B.M.; Dieskau, A.P.; Overman, L.E.; Cotman, C.W. H3K9me3 inhibition improves memory, promotes spine formation, and increases BDNF levels in the aged hippocampus. J. Neurosci. 2016, 36, 3611–3622. [Google Scholar] [CrossRef] [Green Version]
- Ting, J.H.; Marks, D.R.; Schleidt, S.S.; Wu, J.N.; Zyskind, J.W.; Lindl, K.A.; Blendy, J.A.; Pierce, R.C.; Jordan-Sciutto, K.L. Targeted gene mutation of E2F1 evokes age-dependent synaptic disruption and behavioral deficits. J. Neurochem. 2014, 129, 850. [Google Scholar] [CrossRef] [Green Version]
- López-Sánchez, N.; Frade, J.M. [P2–139]: A mutant form of E2F4 prevents neuronal tetraploidization and cognitive deficits in 5xFAD mice without affecting Aβ deposition. Alzheimer’s Dement. 2017, 13, P659–P661. [Google Scholar] [CrossRef]
- Hoerndli, F.J.; Brockie, P.J.; Wang, R.; Mellem, J.E.; Kallarackal, A.; Doser, R.L.; Pierce, D.M.; Madsen, D.M.; Maricq, A.V. MAPK signaling and a mobile scaffold complex regulate AMPA receptor transport to modulate synaptic strength. Cell Rep. 2022, 38, 110577. [Google Scholar] [CrossRef]
- Navarrete, M.; Cuartero, M.I.; Palenzuela, R.; Draffin, J.E.; Konomi, A.; Serra, I.; Colié, S.; Castaño-Castaño, S.; Hasan, M.T.; Nebreda, Á.R.; et al. Astrocytic p38α MAPK drives NMDA receptor-dependent long-term depression and modulates long-term memory. Nat. Commun. 2019, 10, 2968. [Google Scholar] [CrossRef] [Green Version]
- Colié, S.; Sarroca, S.; Palenzuela, R.; Garcia, I.; Matheu, A.; Corpas, R.; Dotti, C.G.; Esteban, J.A.; Sanfeliu, C.; Nebreda, A.R. Neuronal p38α mediates synaptic and cognitive dysfunction in an Alzheimer’s mouse model by controlling β-amyloid production. Sci. Rep. 2017, 7, 45306. [Google Scholar] [CrossRef]
- Schnöder, L.; Gasparoni, G.; Nordström, K.; Schottek, A.; Tomic, I.; Christmann, A.; Schäfer, K.H.; Menger, M.D.; Walter, J.; Fassbender, K.; et al. Neuronal deficiency of p38α-MAPK ameliorates symptoms and pathology of APP or Tau-transgenic Alzheimer’s mouse models. FASEB J. 2020, 34, 9628–9649. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimer’s Dis. 2001, 3, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Misner, D.L.; Sullivan, J.M. Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J. Neurosci. 1999, 19, 6795–6805. [Google Scholar] [CrossRef] [Green Version]
- Navarrete, M.; Araque, A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 2010, 68, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Carlson, G.; Wang, Y.; Alger, B.E. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat. Neurosci. 2002, 5, 723–724. [Google Scholar] [CrossRef] [PubMed]
- Madroñal, N.; Gruart, A.; Valverde, O.; Espadas, I.; Moratalla, R.; Delgado-García, J.M. Involvement of cannabinoid CB1 receptor in associative learning and in hippocampal CA3-CA1 synaptic plasticity. Cereb. Cortex 2012, 22, 550–566. [Google Scholar] [CrossRef] [Green Version]
- Robin, L.M.; Oliveira da Cruz, J.F.; Langlais, V.C.; Martin-Fernandez, M.; Metna-Laurent, M.; Busquets-Garcia, A.; Bellocchio, L.; Soria-Gomez, E.; Papouin, T.; Varilh, M.; et al. Astroglial CB1 Receptors Determine Synaptic D-Serine Availability to Enable Recognition Memory. Neuron 2018, 98, e5–e944. [Google Scholar] [CrossRef] [Green Version]
- Auclair, N.; Otani, S.; Soubrie, P.; Crepelm, F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J. Neurophysiol. 2000, 83, 3287–3293. [Google Scholar] [CrossRef]
- Regehr, W.G.; Tank, D.W. Postsynaptic NMDA receptor-mediated calcium accumulation in hippocampal CA1 pyramidal cell dendrites. Nature 1990, 345, 807–810. [Google Scholar] [CrossRef]
- Hayashi, Y.; Shi, S.H.; Esteban, J.A.; Piccini, A.; Poncer, J.C.; Malinow, R. Driving AMPA receptors into synapses by LTP and CaMKII: Requirement for GluR1 and PDZ domain interaction. Science 2000, 287, 2262–2267. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.J.; Stevens, C.; Tonegawa, S.; Wang, Y. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science 1992, 257, 201–206. [Google Scholar] [CrossRef]
- Chang, J.Y.; Parra-Bueno, P.; Laviv, T.; Szatmari, E.M.; Lee, S.J.R.; Yasuda, R. CaMKII Autophosphorylation Is Necessary for Optimal Integration of Ca2+ Signals during LTP Induction, but Not Maintenance. Neuron 2017, 94, 800–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 2011, 472, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, J. Selecting a mouse model of Alzheimer’s disease. Methods Mol. Biol. 2011, 670, 169–189. [Google Scholar] [CrossRef] [PubMed]
- Ameen-Ali, K.E.; Wharton, S.B.; Simpson, J.E.; Heath, P.R.; Sharp, P.; Berwick, J. Review: Neuropathology and behavioural features of transgenic murine models of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 553–570. [Google Scholar] [CrossRef]
- Amram, S.; Frenkel, D. Animal Models of Alzheimer’s Disease. In Neuroprotection in Alzheimer’s Disease; Gozes, I., Ed.; Elsevier: London, UK, 2017. [Google Scholar]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [CrossRef]
- Clare, R.; King, V.G.; Wirenfeldt, M.; Vinters, H.V. Synapse loss in dementias. J. Neurosci. Res. 2010, 88, 2083–2090. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Hetz, C. Glucose Metabolism: A Sweet Relief of Alzheimer’s Disease. Curr. Biol. 2016, 26, R806–R809. [Google Scholar] [CrossRef] [Green Version]
- Tonnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R. The pre-capillary segment of the blood-brain barrier and its relation to perivascular drainage in Alzheimer’s disease and small vessel disease. Sci. World J. 2009, 9, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar] [CrossRef] [Green Version]
- Frade, J.M.; Ovejero-Benito, M.C. Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle 2015, 14, 712–720. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial hypothesis and multi-targets for Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Kong, W.; Mou, X.; Zhi, X.; Zhang, X.; Yang, Y. Dynamic regulatory network reconstruction for Alzheimer’s disease based on matrix decomposition techniques. Comput. Math. Methods Med. 2014, 2014, 891761. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, A.B.; Liu, Q.; Schroth, G.P.; Galasko, D.R.; Yuan, S.H.; Wagner, S.L.; Subramaniam, S. Dedifferentiation and neuronal repression define familial Alzheimer’s disease. Sci. Adv. 2020, 6, eaba5933. [Google Scholar] [CrossRef]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function. J. Alzheimer’s Dis. 2019, 68, 991–1011. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Ezerskiy, L.A.; Bertelsen, S.; Alzheimer’s Disease Genetics Consortium (ADGC); Goate, A.M. Alzheimer’s Disease Risk Polymorphisms Regulate Gene Expression in the ZCWPW1 and the CELF1 Loci. PLoS ONE 2016, 11, e0148717. [Google Scholar] [CrossRef] [Green Version]
- Morillo, S.M.; Escoll, P.; de la Hera, A.; Frade, J.M. Somatic tetraploidy in specific chick retinal ganglion cells induced by nerve growth factor. Proc. Natl. Acad. Sci. USA 2010, 107, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Sergi, G.; De Rui, M.; Coin, A.; Inelmen, E.M.; Manzato, E. Weight loss and Alzheimer’s disease: Temporal and aetiologic connections. Proc. Nutr. Soc. 2013, 72, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Kagias, K.; Nehammer, C.; Pocock, R. Neuronal responses to physiological stress. Front. Genet. 2012, 3, 222. [Google Scholar] [CrossRef] [Green Version]
- Busser, J.; Geldmacher, D.S.; Herrup, K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J. Neurosci. 1998, 18, 2801–2807. [Google Scholar] [CrossRef] [Green Version]
- Hoozemans, J.J.; Brückner, M.K.; Rozemuller, A.J.; Veerhuis, R.; Eikelenboom, P.; Arendt, T. Cyclin D1 and cyclin E are co-localized with cyclo-oxygenase 2 (COX-2) in pyramidal neurons in Alzheimer disease temporal cortex. J. Neuropathol. Exp. Neurol. 2002, 61, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Vincent, I.; Bu, B.; Hudson, K.; Husseman, J.; Nochlin, D.; Jin, L. Constitutive Cdc25B tyrosine phosphatase activity in adult brain neurons with M phase-type alterations in Alzheimer’s disease. Neuroscience 2001, 105, 639–650. [Google Scholar] [CrossRef]
- Jordan-Sciutto, K.L.; Malaiyandi, L.M.; Bowser, R. Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J. Neuropathol. Exp. Neurol. 2002, 61, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.; Siedlak, S.L.; James, S.L.; Bonda, D.J.; Rao, A.; Webber, K.M.; Camins, A.; Pallàs, M.; Casadesus, G.; Lee, H.G.; et al. Retinoblastoma protein phosphorylation at multiple sites is associated with neurofibrillary pathology in Alzheimer disease. Int. J. Clin. Exp. Pathol. 2008, 1, 134. [Google Scholar]
- Nagy, Z.; Esiri, M.M.; Cato, A.M.; Smith, A.D. Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol. 1997, 94, 6–15. [Google Scholar] [CrossRef]
- Giovanni, A.; Wirtz-Brugger, F.; Keramaris, E.; Slack, R.; Park, D.S. Involvement of Cell Cycle Elements, Cyclin-dependent Kinases, pRb, and E2F·DP, in B-amyloid-induced Neuronal Death. J. Biol. Chem. 1999, 274, 19011–19016. [Google Scholar] [CrossRef] [Green Version]
- Park, K.H.J.; Hallows, J.L.; Chakrabarty, P.; Davies, P.; Vincent, I. Conditional neuronal simian virus 40 T antigen expression induces Alzheimer-like tau and amyloid pathology in mice. J. Neurosci. 2007, 27, 2969–2978. [Google Scholar] [CrossRef] [Green Version]
- McShea, A.; Lee, H.G.; Petersen, R.B.; Casadesus, G.; Vincent, I.; Linford, N.J.; Funk, J.O.; Shapiro, R.A.; Smith, M.A. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim. Biophys. Acta 2007, 1772, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.G.; Casadesus, G.; Nunomura, A.; Zhu, X.; Castellani, R.J.; Richardson, S.L.; Perry, G.; Felsher, D.W.; Petersen, R.B.; Smith, M.A. The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am. J. Pathol. 2009, 174, 891–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.H.J.; Barrett, T. Gliosis precedes amyloid-β deposition and pathological tau accumulation in the neuronal cell cycle re-entry mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. Rep. 2020, 4, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Barrio-Alonso, E.; Hernández-Vivanco, A.; Walton, C.C.; Perea, G.; Frade, J.M. Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci. Rep. 2018, 8, 14316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Stangis, K.A.; Saito, T.; Saido, T.; Park, K.H.J. Neuronal Cell Cycle Re-Entry Enhances Neuropathological Features in AppNLF Knock-In Mice. J. Alzheimer’s Dis. 2021, 82, 1683–1702. [Google Scholar] [CrossRef]
- Yang, Y.; Geldmacher, D.S.; Herrup, K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 2001, 21, 2661–2668. [Google Scholar] [CrossRef] [Green Version]
- Mosch, B.; Morawski, M.; Mittag, A.; Lenz, D.; Tarnok, A.; Arendt, T. Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J. Neurosci. 2007, 27, 6859–6867. [Google Scholar] [CrossRef]
- López-Sánchez, N.; Fontán-Lozano, Á.; Pallé, A.; González-Álvarez, V.; Rábano, A.; Trejo, J.L.; Frade, J.M. Neuronal tetraploidization in the cerebral cortex correlates with reduced cognition in mice and precedes and recapitulates Alzheimer’s-associated neuropathology. Neurobiol. Aging 2017, 56, 50–66. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Mosch, B.; Lösche, A. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 2010, 177, 15–20. [Google Scholar] [CrossRef]
- Arendt, T. Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 125–135. [Google Scholar] [CrossRef]
- Barrio-Alonso, E.; Fontana, B.; Valero, M.; Frade, J.M. Pathological aspects of neuronal hyperploidization in Alzheimer’s disease evidenced by computer simulation. Front. Genet. 2020, 11, 287. [Google Scholar] [CrossRef]
- Murman, D.L. The Impact of Age on Cognition. Semin. Hear. 2015, 36, 111–121. [Google Scholar] [CrossRef]
- Roesler, R.; McGaugh, J.L. The Entorhinal Cortex as a Gateway for Amygdala Influences on Memory Consolidation. Neuroscience 2022, 497, 86–96. [Google Scholar] [CrossRef]
- Simon, E.; Obst, J.; Gomez-Nicola, D. The evolving dialogue of microglia and neurons in Alzheimer’s disease: Microglia as necessary transducers of pathology. Neuroscience 2019, 405, 24–34. [Google Scholar] [CrossRef]
- Bahrini, I.; Song, J.H.; Diez, D.; Hanayama, R. Neuronal exosomes facilitate synaptic pruning by up-regulating complement factors in microglia. Sci. Rep. 2015, 5, 7989. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.N.; Yang, C.L.; Lin, K.N.; Chen, W.T.; Chwang, L.C.; Liu, H.C. Weight loss, nutritional status and physical activity in patients with Alzheimer’s disease. A controlled study. J. Neurol. 2004, 251, 314–320. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Serraneau, K.S.; Brownlow, M.; Marín de Evsikova, C.; Speakman, J.R.; Gordon, M.N.; Morgan, D. Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging 2016, 44, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Wang, G.; Racchumi, G.; Dyke, J.P.; Iadecola, C. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. 2014, 34, 9096–9106. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- Arendt, T. Synaptic plasticity and cell cycle activation in neurons are alternative effector pathways: The ‘Dr. Jekyll and Mr. Hyde concept’ of Alzheimer’s disease or the yin and yang of neuroplasticity. Prog. Neurobiol. 2003, 71, 83–248. [Google Scholar] [CrossRef]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705–716. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, A.K.Y.; Ip, N.Y. Instructive roles of astrocytes in hippocampal synaptic plasticity: Neuronal activity-dependent regulatory mechanisms. FEBS J. 2022, 289, 2202–2218. [Google Scholar] [CrossRef]
- Sciaccaluga, M.; Megaro, A.; Bellomo, G.; Ruffolo, G.; Romoli, M.; Palma, E.; Costa, C. An Unbalanced Synaptic Transmission: Cause or Consequence of the Amyloid Oligomers Neurotoxicity? Int. J. Mol. Sci. 2021, 22, 5991. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.L.; Meyer, M.; Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001, 4, 29–37. [Google Scholar] [CrossRef]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia Priming with Aging and stress. Neuropsychopharmacology 2017, 42, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J.R. Microglia in aging and Alzheimer’s disease: A comparative species review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef]
- Reeves, S.; Helman, L.; Allison, A.; Israel, M. Molecular cloning and primary structure of human glial fibrillary acidic protein. Proc. Natl. Acad. Sci. USA 1989, 86, 5178–5182. [Google Scholar] [CrossRef] [Green Version]
- Nathwani, A.C.; Reiss, U.; Tuddenham, E.; Chowdary, P.; McIntosh, J.; Riddell, A.; Pie, J.; Mahlangu, J.N.; Recht, M.; Shen, Y.-M.; et al. Adeno-Associated Mediated Gene Transfer for Hemophilia B:8 Year Follow up and Impact of Removing “Empty Viral Particles” on Safety and Efficacy of Gene Transfer. Blood 2018, 132 (Suppl. S1), 491. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Héraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucl. Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramón-Landreau, M.; Sánchez-Puelles, C.; López-Sánchez, N.; Lozano-Ureña, A.; Llabrés-Mas, A.M.; Frade, J.M. E2F4DN Transgenic Mice: A Tool for the Evaluation of E2F4 as a Therapeutic Target in Neuropathology and Brain Aging. Int. J. Mol. Sci. 2022, 23, 12093. https://doi.org/10.3390/ijms232012093
Ramón-Landreau M, Sánchez-Puelles C, López-Sánchez N, Lozano-Ureña A, Llabrés-Mas AM, Frade JM. E2F4DN Transgenic Mice: A Tool for the Evaluation of E2F4 as a Therapeutic Target in Neuropathology and Brain Aging. International Journal of Molecular Sciences. 2022; 23(20):12093. https://doi.org/10.3390/ijms232012093
Chicago/Turabian StyleRamón-Landreau, Morgan, Cristina Sánchez-Puelles, Noelia López-Sánchez, Anna Lozano-Ureña, Aina M. Llabrés-Mas, and José M. Frade. 2022. "E2F4DN Transgenic Mice: A Tool for the Evaluation of E2F4 as a Therapeutic Target in Neuropathology and Brain Aging" International Journal of Molecular Sciences 23, no. 20: 12093. https://doi.org/10.3390/ijms232012093
APA StyleRamón-Landreau, M., Sánchez-Puelles, C., López-Sánchez, N., Lozano-Ureña, A., Llabrés-Mas, A. M., & Frade, J. M. (2022). E2F4DN Transgenic Mice: A Tool for the Evaluation of E2F4 as a Therapeutic Target in Neuropathology and Brain Aging. International Journal of Molecular Sciences, 23(20), 12093. https://doi.org/10.3390/ijms232012093