
Checkpoint Kinase 2 Inhibition Can Reverse Tamoxifen Resistance in ER-Positive Breast Cancer

 , and
, and
Abstract
:1. Introduction
2. Results
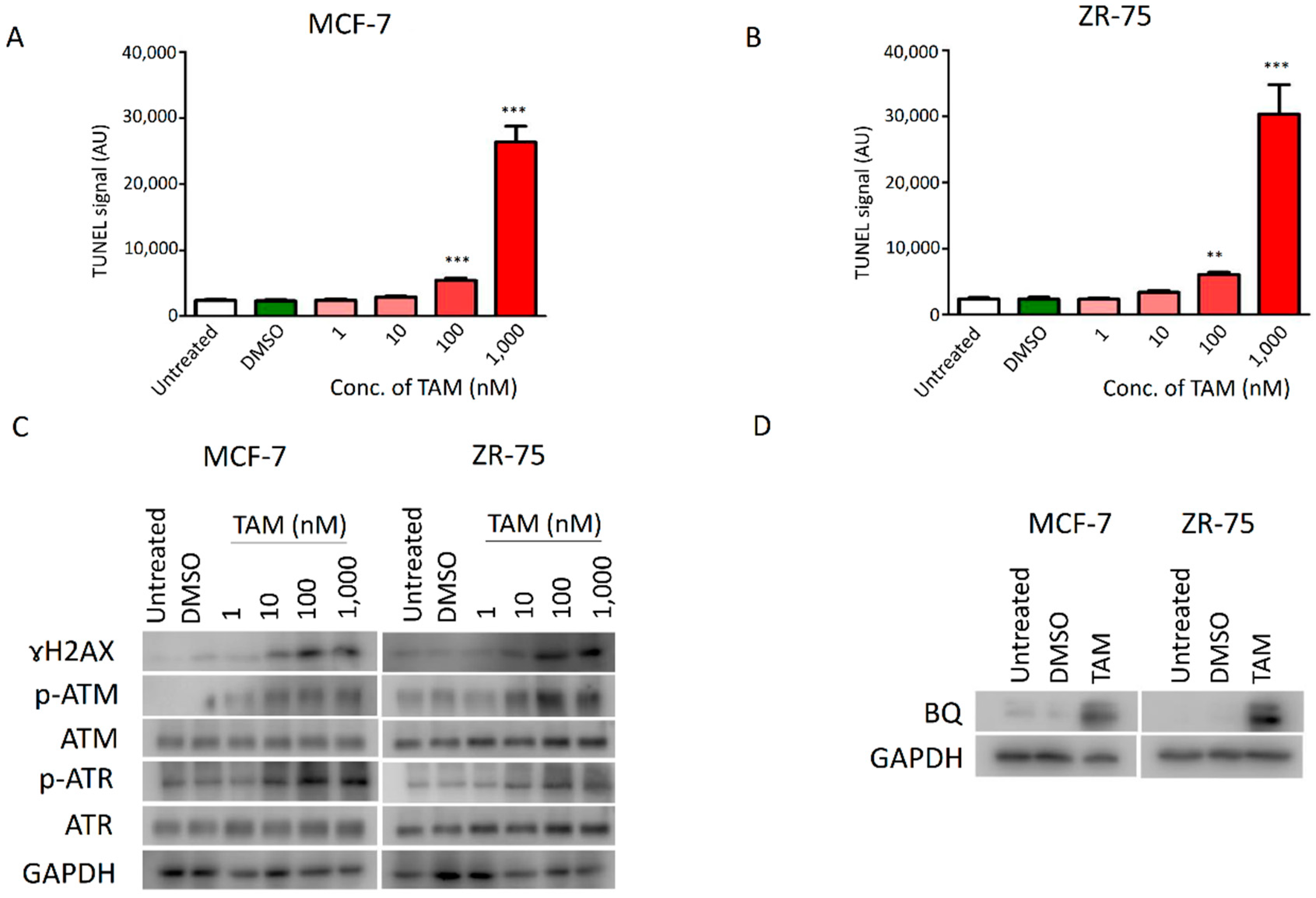
2.1. DNA Damage Response (DDR) Signalling Modulated the Expression of BQ in Breast Cancer
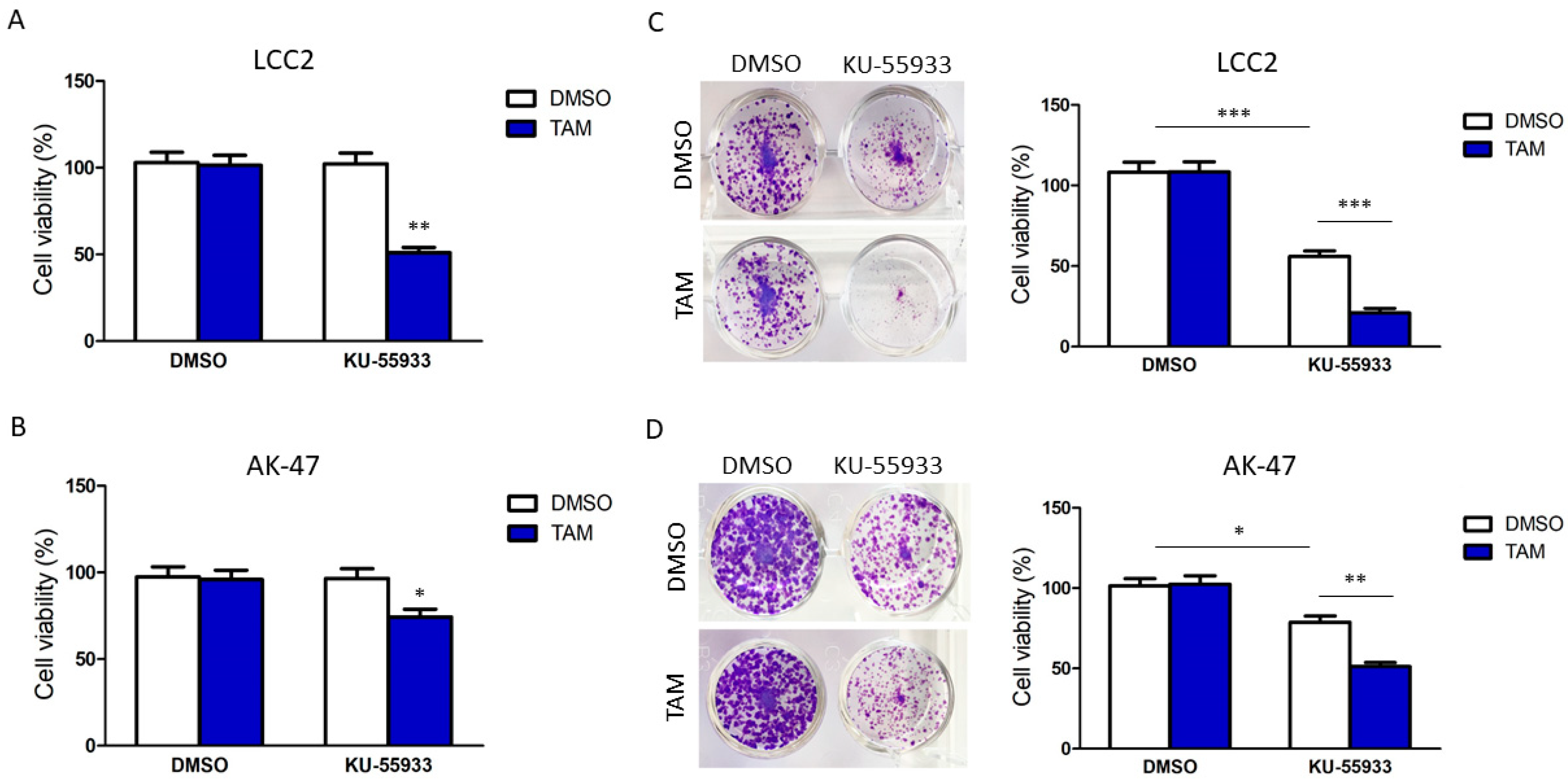
2.2. Suppression of CHK2 Activity Could Reduce Expression of BQ and Thus Tamoxifen Resistance
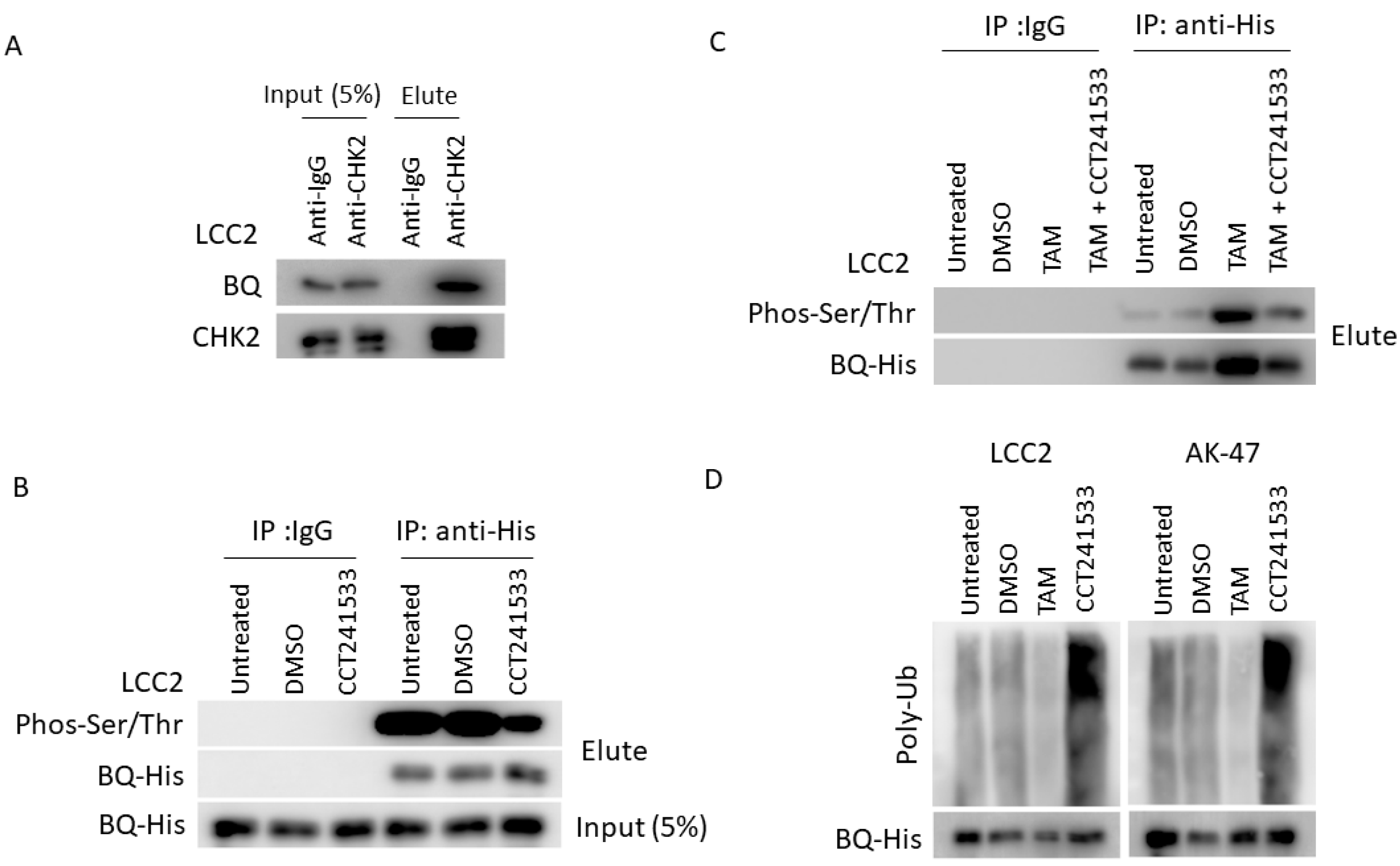
2.3. CHK2 Could Modulate the Protein Stability of BQ in Breast Cancer Cells
2.4. Clinical Significance of p-CHK2 in ER + ve Breast Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Transfection and Stable Cell Line Establishment
4.2. Plasmids, siRNA, shRNA and RT-qPCR
4.3. Western Blot
4.4. Co-Immunoprecipitation
4.5. Cell Viability and Functional Assays
4.6. Chemicals
4.7. Xenograft
4.8. Tissue Microarray and Immunohistochemistry (IHC)
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. Cancer J. Clin 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Therapeut. 2018, 186, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Darby, S.; McGale, P.; Wang, Y.C.; Peto, R.; Pan, H.C.; Cutter, D.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Riggins, R.B.; Schrecengost, R.S.; Guerrero, M.S.; Bouton, A.H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Sette, C.; Ladomery, M.; Ghigna, C. Alternative splicing: Role in cancer development and progression. Int. J. Cell Biol. 2013, 2013, 421606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.D.; Gong, C.; Lau, S.L.Y.; Yang, N.; Wong, O.G.W.; Cheung, A.N.Y.; Tsang, J.W.H.; Chan, K.Y.K.; Khoo, U.S. SpliceArray Profiling of Breast Cancer Reveals a Novel Variant of NCOR2/SMRT That Is Associated with Tamoxifen Resistance and Control of ER alpha Transcriptional Activity. Cancer Res. 2013, 73, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, H.; Man, E.P.; Leung, M.H.; Mok, K.C.; Chau, K.M.; Wong, L.S.; Chan, W.L.; Chan, S.Y.; Luk, M.Y.; Cheng, C.N.; et al. KPNA1 regulates nuclear import of NCOR2 splice variant BQ323636.1 to confer tamoxifen resistance in breast cancer. Clin. Transl. Med. 2021, 11, e554. [Google Scholar] [CrossRef]
- Tsoi, H.; Shi, L.; Leung, M.-H.; Man, E.P.S.; So, Z.-Q.; Chan, W.-L.; Khoo, U.-S. Overexpression of BQ323636.1 Modulated AR/IL-8/CXCR1 Axis to Confer Tamoxifen Resistance in ER-Positive Breast Cancer. Life 2022, 12, 93. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Samec, M.; Liskova, A.; Kubatka, P.; Saso, L. Tamoxifen and oxidative stress: An overlooked connection. Discov. Oncol. 2021, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol 2019, 25, 101084. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Liu, Y.J.; Zhang, C.; Chu, J.J.; Wu, Y.Q.; Li, Y.D.; Liu, J.Q.; Li, Q.; Li, S.Y.; Shi, Q.F.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9, 1595. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Man, E.P.S.; Tsoi, H.; Lee, T.K.W.; Lee, P.; Ma, S.T.; Wong, L.S.; Luk, M.Y.; Rakha, E.A.; Green, A.R.; et al. BQ323636.1, a Novel Splice Variant to NCOR2, as a Predictor for Tamoxifen-Resistant Breast Cancer. Clin. Cancer Res. 2018, 24, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, M.H.; Tsoi, H.; Gong, C.; Man, E.P.S.; Zona, S.; Yao, S.; Lam, E.W.F.; Khoo, U.S. A Splice Variant of NCOR2, BQ323636.1, Confers Chemoresistance in Breast Cancer by Altering the Activity of NRF2. Cancers 2020, 12, 533. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell. Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [Green Version]
- Caldoe, C.E. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front. Oncol 2014, 4, 106. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Griesbach, E.; Schlackow, M.; Marzluff, W.F.; Proudfoot, N.J. Dual RNA 3′-end processing of H2A.X messenger RNA maintains DNA damage repair throughout the cell cycle. Nat. Commun. 2021, 12, 359. [Google Scholar] [CrossRef]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 Is a Potent and Selective Inhibitor of CHK2 that Potentiates the Cytotoxicity of PARP Inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular Inhibition of Checkpoint Kinase 2 (Chk2) and Potentiation of Camptothecins and Radiation by the Novel Chk2 Inhibitor PV1019 [7-Nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharmacol. Exp. Ther 2009, 331, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the Understanding of the Mechanism of Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef]
- Rasha, F.; Sharma, M.; Pruitt, K. Mechanisms of endocrine therapy resistance in breast cancer. Mol. Cell. Endocrinol. 2021, 532, 111322. [Google Scholar] [CrossRef]
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, P.Z.; Zhang, W.; Zhang, X.; Chen, J.F.; Ding, P.P.; Li, L.Y.; Lv, X.Y.; Li, L.; Hu, W.G. PI3K activation is enhanced by FOXM1D binding to p110 and p85 subunits. Signal Transduct. Target. Ther. 2020, 5, 105. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.W.; Liu, G.Z.; Li, C.X.; Song, Y.R.; Cao, Z.W.; Li, W.; Hu, J.H.; Lu, C.; Liu, Y.Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Hanamura, T.; Hayashi, S. Overcoming aromatase inhibitor resistance in breast cancer: Possible mechanisms and clinical applications. Breast Cancer 2018, 25, 379–391. [Google Scholar] [CrossRef]
- Sabine, V.S.; Crozier, C.; Brookes, C.L.; Drake, C.; Piper, T.; van de Velde, C.J.H.; Hasenburg, A.; Kieback, D.G.; Markopoulos, C.; Dirix, L.; et al. Mutational Analysis of PI3K/AKT Signaling Pathway in Tamoxifen Exemestane Adjuvant Multinational Pathology Study. J. Clin. Oncol. 2014, 32, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Man, E.P.S.; Chau, K.M.; Khoo, U.S. Targeting the IL-6/STAT3 Signalling Cascade to Reverse Tamoxifen Resistance in Estrogen Receptor Positive Breast Cancer. Cancers 2021, 13, 1511. [Google Scholar] [CrossRef]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Xu, L.; Wang, P.; Hu, X.; Zhang, R.; Wu, Z.; Du, W.; Kan, W.; Li, K.; Wang, C.; et al. Discovery and Development of a Potent, Selective, and Orally Bioavailable CHK1 Inhibitor Candidate: 5-((4-((3-Amino-3-methylbutyl)amino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)picolinonitrile. J. Med. Chem. 2021, 64, 15069–15090. [Google Scholar] [CrossRef]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Nishi, H.; Shaytan, A.; Panchenko, A.R. Physicochemical mechanisms of protein regulation by phosphorylation. Front. Genet. 2014, 5, 270. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.Y.; Kuo, I.Y.; Chen, Y.T.; Liao, P.C.; Liu, Y.F.; Wu, H.Y.; Lai, W.W.; Wang, Y.C. AKT-mediated phosphorylation enhances protein stability and transcription activity of ZNF322A to promote lung cancer progression. Oncogene 2019, 38, 6723–6736. [Google Scholar] [CrossRef]
- Liu, H.; Wang, K.T.; Chen, S.; Sun, Q.; Zhang, Y.K.; Chen, L.; Sun, X.L. NFATc1 phosphorylation by DYRK1A increases its protein stability. PLoS ONE 2017, 12, e0172985. [Google Scholar] [CrossRef] [Green Version]
- Devaiah, B.N.; Mu, J.; Akman, B.; Uppal, S.; Weissman, J.D.; Cheng, D.; Baranello, L.; Nie, Z.Q.; Levens, D.; Singer, D.S. MYC protein stability is negatively regulated by BRD4. Proc. Natl. Acad. Sci. USA 2020, 117, 13457–13467. [Google Scholar] [CrossRef]
- Huang, Q.L.; Zhang, X.F. Emerging Roles and Research Tools of Atypical Ubiquitination. Proteomics 2020, 20, 1900100. [Google Scholar] [CrossRef]
- Fujimoto, H.; Onishi, N.; Kato, N.; Takekawa, M.; Xu, X.; Kosugi, A.; Kondo, T.; Imamura, M.; Oishi, I.; Yoda, A.; et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006, 13, 1170–1180. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||||
| Overall Survival | Disease-Specific Survival | ||||
| Clinical–Pathological Parameters | Cases | RR (95% CI) | p Value | RR (95% CI) | p Value |
| Age | 183 | 1.994 (1.241, 3.204) | 0.004 | 1.102 (0.618, 1.967) | 0.742 |
| T stage | 133 | 1.579 (0.564, 4.423) | 0.384 | 1.150 (0.273, 4.852) | 0.849 |
| Lymph node involvement | 169 | 1.446 (0.870, 2.404) | 0.155 | 2.148 (1.116, 4.137) | 0.022 |
| Tumour grade | 174 | 1.357 (0.834, 2.208) | 0.220 | 2.200 (1.198, 4.041) | 0.011 |
| Histological type | 183 | 1.020 (0.536, 1.942) | 0.952 | 0.699 (0.347, 1.406) | 0.315 |
| PR status | 169 | 0.686 (0.384, 1.225) | 0.202 | 0.484 (0.251, 0.931) | 0.030 |
| HER2 status | 140 | 1.084 (0.527, 2.226) | 0.827 | 1.114 (0.459, 2.702) | 0.812 |
| Tumour size | 65 | 1.417 (0.581, 3.455) | 0.444 | 2.248 (0.623, 8.110) | 0.216 |
| High nuclear pCHK2 score | 170 | 2.555 (1.526, 4.277) | 3.59 × 10−4 | 2.781 (1.458, 5.305) | 0.002 |
| High nuclear BQ score | 171 | 2.713 (1.620, 4.545) | 1.49 × 10−4 | 2.897 (1.519, 5.527) | 0.001 |
| Both high nuclear pCHK2 and BQ score | 103 | 5.196 (2.395, 11.271) | 3.04 × 10−5 | 4.944 (2.016, 12.127) | 4.81 × 10−4 |
| (B) | |||||
| Overall Survival | Disease-Specific Survival | ||||
| Clinical–Pathological Parameters | Cases | RR (95% CI) | p Value | RR (95% CI) | p Value |
| Age | 170 | 1.919 (1.187, 3.103) | 0.008 | ||
| High nuclear pCHK2 score | 170 | 2.588 (1.545, 4.333) | 3.01 × 10−4 | ||
| Age | 171 | 1.953 (1.208, 3.156) | 0.006 | ||
| High nuclear BQ score | 171 | 2.721 (1.624, 4.559) | 1.43 × 10−4 | ||
| Age | 103 | 1.361 (0.736, 2.518) | 0.326 | ||
| Both high nuclear pCHK2 & BQ score | 103 | 5.181 (2.388, 11.242) | 3.14 × 10−5 | ||
| Lymph node involvement | 142 | 2.509 (1.226, 5.134) | 0.012 | ||
| Tumour grade | 142 | 1.701 (0.845, 3.426) | 0.137 | ||
| PR status | 142 | 0.391 (0.190, 0.805) | 0.011 | ||
| High nuclear pCHK2 score | 142 | 3.344 (1.585, 7.058) | 0.002 | ||
| Lymph-node involvement | 141 | 2.369 (1.133, 4.952) | 0.022 | ||
| Tumour grade | 141 | 1.795 (0.908, 3.549) | 0.093 | ||
| PR status | 141 | 0.384 (0.183, 0.807) | 0.012 | ||
| High nuclear BQ score | 141 | 2.639 (1.286, 5.413) | 0.008 | ||
| Lymph node involvement | 89 | 2.711 (1.112, 6.610) | 0.028 | ||
| Tumour grade | 89 | 1.277 (0.533, 3.060) | 0.583 | ||
| PR status | 89 | 0.441 (0.172, 1.130) | 0.088 | ||
| Both high nuclear pCHK2 and BQ score | 89 | 5.393 (1.961, 14.831) | 0.001 | ||
| Clinical Characters | Number of CASES | Percentage (%) | |
|---|---|---|---|
| Breast cancer patients | 313 | 100 | |
| Age | <54 | 161 | 51.4 |
| ≥54 | 150 | 47.9 | |
| T stage | I, II | 180 | 57.5 |
| III, IV | 17 | 5.4 | |
| Lymph Node status | Positive | 147 | 47.0 |
| Negative | 132 | 42.2 | |
| Tumour Grade | 1, 2 | 131 | 41.9 |
| 3 | 148 | 47.3 | |
| Tumour Size | <2 cm | 39 | 12.5 |
| ≥2 cm | 71 | 22.7 | |
| Oestrogen Receptor status | Positive | 185 | 59.1 |
| Negative | 66 | 21.1 | |
| Progesterone receptor status | Positive | 142 | 45.4 |
| Negative | 95 | 30.4 | |
| HER2 receptor status | Positive | 38 | 12.1 |
| Negative | 158 | 50.5 | |
| Triple Negative status | Positive | 36 | 11.5 |
| Negative | 189 | 60.4 | |
| pCHK2 | No expression | 77 | 24.6 |
| Expression | 236 | 70.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoi, H.; Tsang, W.-C.; Man, E.P.S.; Leung, M.-H.; You, C.-P.; Chan, S.-Y.; Chan, W.-L.; Khoo, U.-S. Checkpoint Kinase 2 Inhibition Can Reverse Tamoxifen Resistance in ER-Positive Breast Cancer. Int. J. Mol. Sci. 2022, 23, 12290. https://doi.org/10.3390/ijms232012290
Tsoi H, Tsang W-C, Man EPS, Leung M-H, You C-P, Chan S-Y, Chan W-L, Khoo U-S. Checkpoint Kinase 2 Inhibition Can Reverse Tamoxifen Resistance in ER-Positive Breast Cancer. International Journal of Molecular Sciences. 2022; 23(20):12290. https://doi.org/10.3390/ijms232012290
Chicago/Turabian StyleTsoi, Ho, Wai-Chung Tsang, Ellen P. S. Man, Man-Hong Leung, Chan-Ping You, Sum-Yin Chan, Wing-Lok Chan, and Ui-Soon Khoo. 2022. "Checkpoint Kinase 2 Inhibition Can Reverse Tamoxifen Resistance in ER-Positive Breast Cancer" International Journal of Molecular Sciences 23, no. 20: 12290. https://doi.org/10.3390/ijms232012290
APA StyleTsoi, H., Tsang, W. -C., Man, E. P. S., Leung, M. -H., You, C. -P., Chan, S. -Y., Chan, W. -L., & Khoo, U. -S. (2022). Checkpoint Kinase 2 Inhibition Can Reverse Tamoxifen Resistance in ER-Positive Breast Cancer. International Journal of Molecular Sciences, 23(20), 12290. https://doi.org/10.3390/ijms232012290