
The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application
Abstract
:
1. Introduction
2. Molecules and Mechanisms of Liver Fibrosis
2.1. Cell Types in Liver Fibrosis
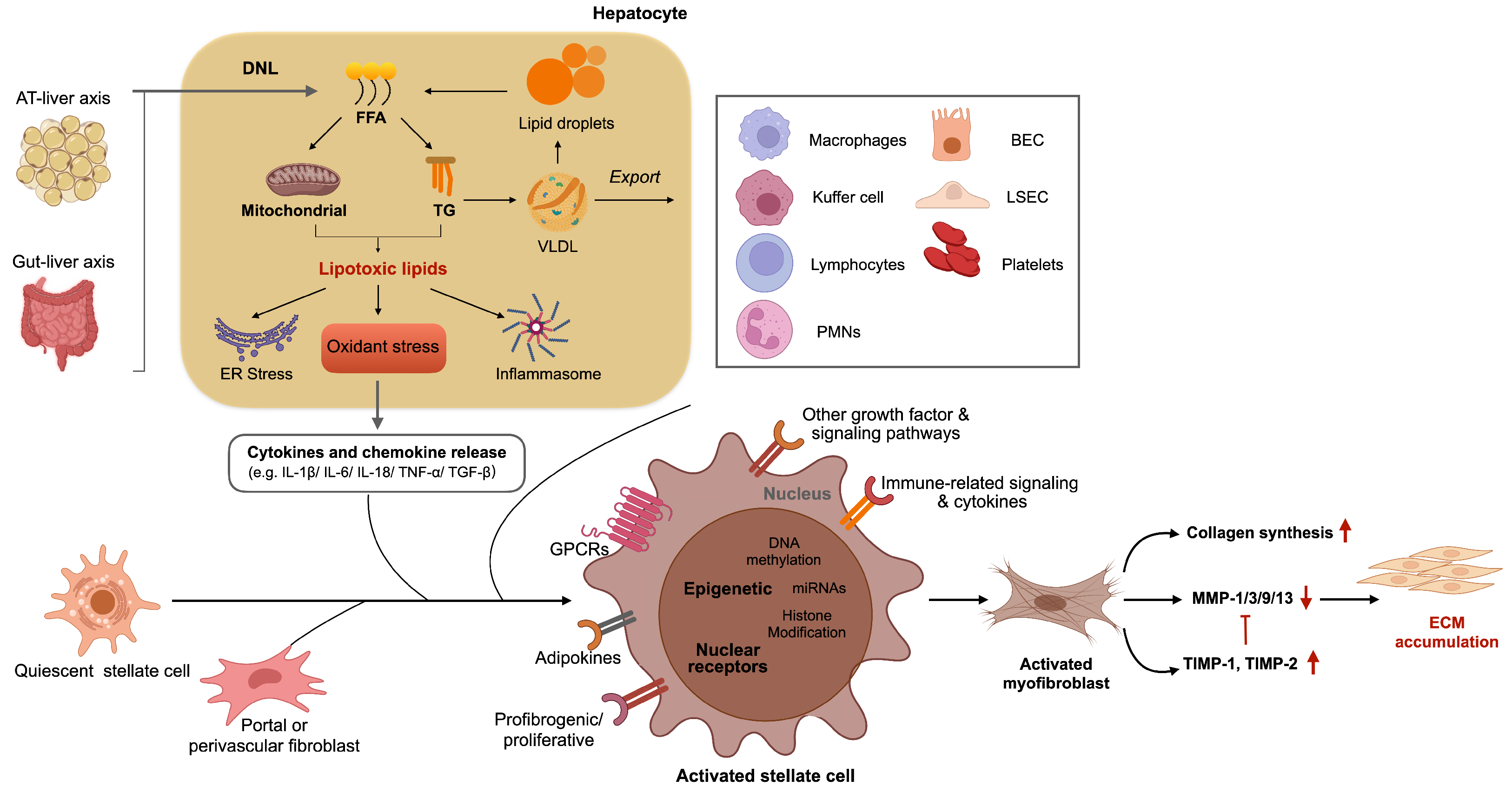
2.1.1. Hepatocytes
2.1.2. Inflammatory Cells
2.1.3. Hepatic Stellate Cell
2.1.4. Myofibroblasts
2.1.5. Liver Sinusoidal Endothelial Cells
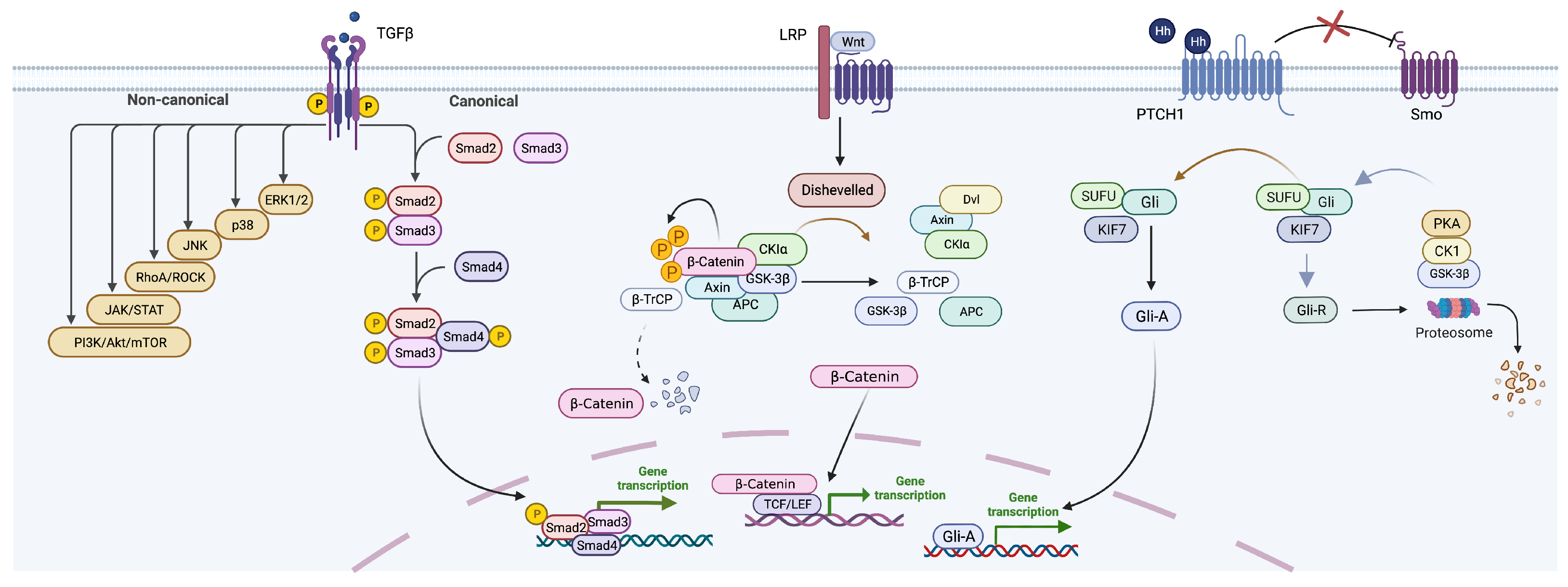
2.2. Key Signaling Pathways in Liver Fibrosis
2.2.1. TGF-b Signaling Pathway
2.2.2. Wnt Signaling Pathway
2.2.3. Hedgehog (Hh) Signaling Pathway
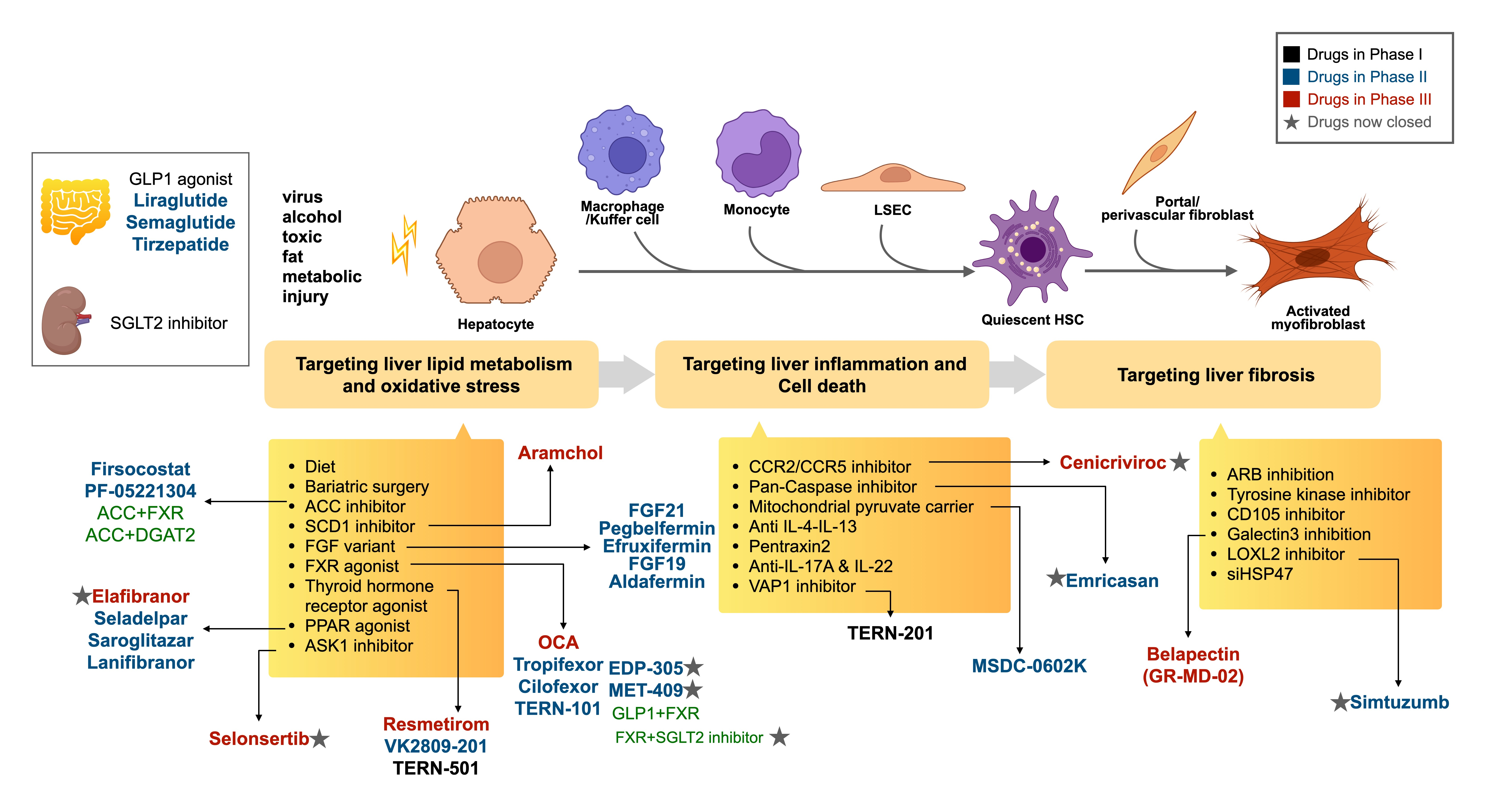
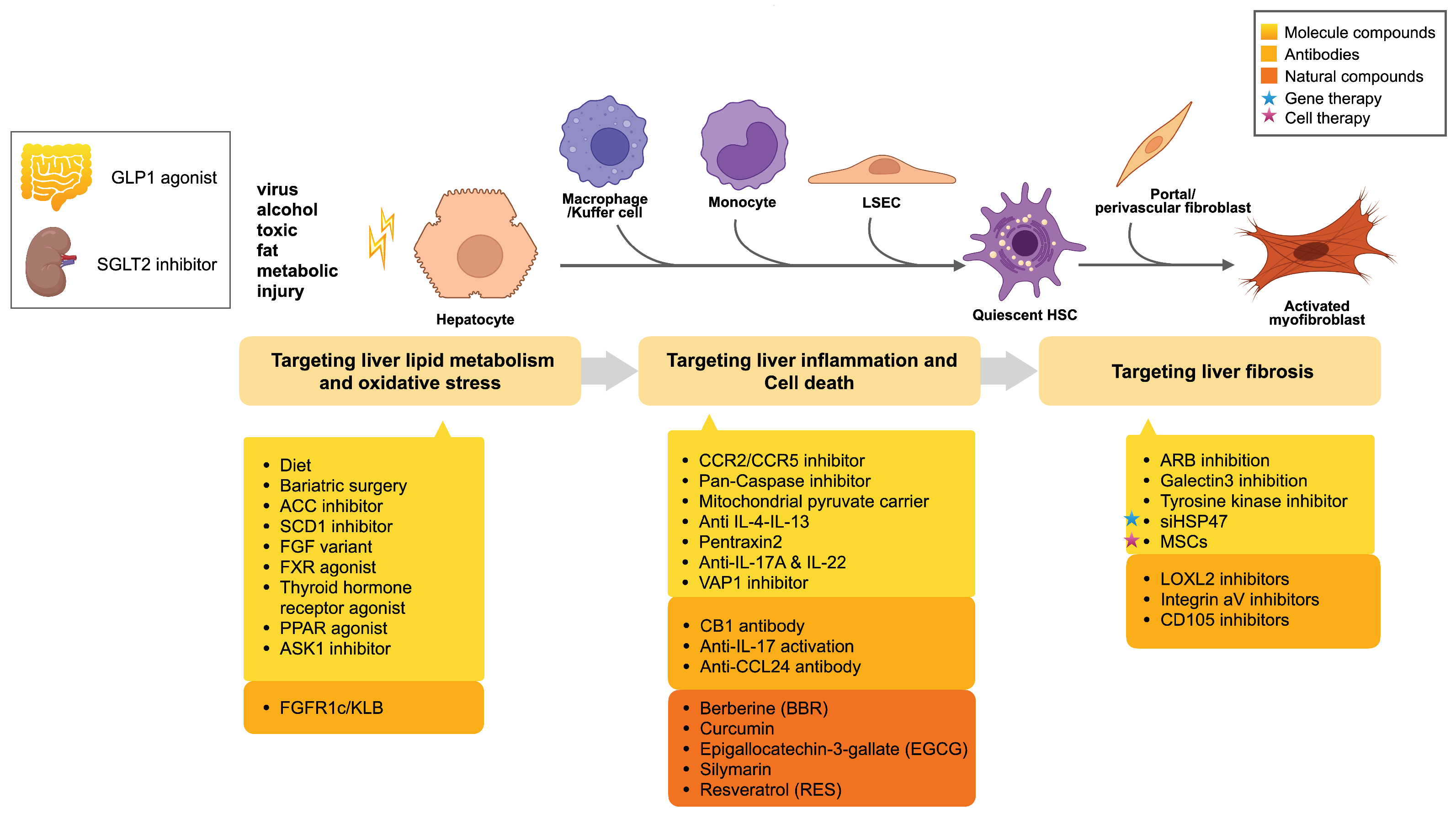
3. Current Interventions in Liver Fibrosis Management
3.1. Pharmacologic Treatments
3.1.1. Targeting Liver Lipid Metabolism and Oxidative Stress
Molecule Compounds
- (1)
- ACC InhibitorsACC (acetyl CoA carboxylase) is a key enzyme regulating lipid metabolism. ACC comes in two informs, which present in different tissues: ACC1 is more highly expressed in liver and adipose tissue, whereas ACC2 is more expressed in oxidative tissues including cardiac muscle and skeletal muscle. Firsocostat (GS-0976), an ACC1 inhibitor, reduced de novo lipogenesis and liver fat in 126 patients with nonalcoholic steatohepatitis (NASH) without cirrhosis but historical biopsy consistent with NASH and F1–F3 fibrosis (NCT02856555). The administration of 20 mg GS-0976 for 12 weeks was shown to be safe, but there was a 13% increase in serum triglycerides, with hypertriglyceridemia being the most common AE (adverse event) [27].
- (2)
- SCD1 inhibitorsThe stearoyl-CoA desaturase 1 (SCD1) enzyme is a rate-limiting enzyme that regulates the monounsaturated fatty acid production process. The inhibition of SCD1 reduces fatty acid synthesis while increasing b-oxidation, resulting in lower hepatic triglycerides. Aramchol, a partial inhibitor of SCD1, forms a stable amide link between two natural components, cholic acid (bile acid) and arachidic acid (saturated fatty acid). NASH resolution without worsening fibrosis was obtained in 16.7% of Aramchol 600 mg against 5% of the placebo arm in a Phase IIb ARREST study NCT02279524, although the primary end point of a reduction in liver fat content with Aramchol 600 mg did not meet the expected significance level. The observed safety and changes in liver histology and enzymes provide a rationale for SCD1 modulation as a promising therapy for NASH and fibrosis and are currently being evaluated in a Phase 3 study [28].
- (3)
- FGF agonistsFibroblast growth factor functions as regulating bile acid synthesis and glucose homeostasis. NGM282 (aldafermin) is a FGF19 analogue. Patients were randomly assigned to receive 3 mg, 6 mg NGM282, or placebo in a study of 82 patients with NASH (NCT02443116) [29]. The absolute change in liver fat content from baseline to week 12 was the primary outcome. The 12 weeks of NGM282 treatment resulted in relative decreases in corrected T1 (cT1; 8% and 9%), ALT (67% and 60%), AST (57% and 52%), and Pro-C3 (22% and 33%). Aldafermin-related AEs were few, mild to moderate, and gastrointestinal in origin [30].Pegbelfermin(BMS-986936) is a pegylated FGF21 analog. The safety, pharmacokinetics, and pharmacodynamic effects of BMS-986036 in 184 people with NASH were evaluated in a 16-week randomized, double-blind trial (NCT02413372) [31]. The group receiving 10 mg pegbelfermin daily (68% vs. 13%; p = 0.0004) and the group receiving 20 mg pegbelfermin weekly (52% vs. 13%; p = 0.008) showed a substantial decrease in absolute hepatic fat fraction compared with the placebo group at week 16. Most AEs were mild [32].
- (4)
- THR beta agonistsThyroid hormone acts as a ligand for two receptors, thyroid hormone receptor-a (THRa) and thyroid hormone receptor-b (THRb). THRa is highly expressed in the heart and bone, whereas THRb is the primary form expressed in the liver, which regulates several processes involved in hepatic triglyceride and cholesterol metabolism to lower serum cholesterol and intrahepatic lipid content [33]. Low thyroid function was associated with a higher prevalence of NASH and advanced fibrosis than strict-normal thyroid function [34].Resmetirom and VK2809 are two orally active THR agonists that are liver-directed and have a several-fold higher selectivity for THRb than THRa. A Phase 2, randomized, double-blind, placebo-controlled study evaluated the effect of MGL-3196 on patients with NASH (fibrosis Stages 1–3) [35]. Patients receiving resmetirom showed a relative reduction of hepatic fat compared with placebo at week 36 (−37.3% versus −8.5; p < 0.0001). AEs were largely mild or moderate and were evenly distributed between groups [36]. Further studies of resmetirom will allow assessment of the safety and effectiveness of resmetirom in a larger number of patients with NASH with the possibility of documenting associations between histological effects and changes in imaging. Resmetirom has a larger total fat-burning effect than the other targets we discussed above, and the most recent Phase 3 results show a positive result in NAFLD. So, it is promising to be the first FDA-approved treatment for NASH. One of the disadvantages of resmetirom is that the metabolized product is a stable and active product, which means that the drug must be adjusted twice to account for individual metabolic differences, which might be inconvenient to use therapeutically.
- (5)
- FXR agonistsThe nuclear receptor FXR (farnesoid X receptor) is mostly expressed in the liver, gallbladder, and intestine. Bile acids, which regulate lipid metabolism and glucose homeostasis, increase insulin sensitivity, and have anti-inflammatory and anti-fibrosis properties, are the natural ligands of FXR [37].Obeticholic acid (OCA) is the most advanced drug in FXR agonists. In a Phase III REGENERATE trial (NCT02548351), 1968 patients with definite NASH were randomly assigned to receive OCA at 10 mg or 25 mg daily or placebo [38]. After 18 months of treatment, the fibrosis improvement end point was achieved by 23% in the OCA 25 mg group (p = 0.0002) versus 12% of patients in the placebo group. Pruritus was the most common AE. Patients with OCA therapy were associated with an increase at 12 weeks in small very low-density lipoprotein (VLDL) and LDL and a reduction in high-density lipoprotein (HDL) [39]. In conclusion, the role of OCA in anti-fibrosis was present, but its overall effectiveness was only about 25%, suggesting that only one out of every four people responds [40]. At the same time, the AEs were rather strong, including 50% pruritus and an increase in LDL, both of which are not good for people with cardiovascular disease [41]. Intercept will resubmit the NDC with 500 additional samples, and the patients in the group have also been observed for a longer period of time, although the specific outcomes have yet to be revealed.Cilofexor (GS-9674) is a selective nonsteroidal FXR agonist that showed promise in a Phase II trial in NASH patients. In this double-blind, placebo-controlled trial, 140 patients with NASH without cirrhosis were randomized to receive cilofexor 100 mg, 30 mg, or placebo orally once daily for 24 weeks (NCT02854605) [42]. Cilofexor was generally well tolerated. Moderate to severe pruritus was more common in patients receiving cilofexor 100 mg (14%) than in those receiving cilofexor 30 mg (4%) and placebo (4%).
- (6)
- PPAR agonistsPeroxisomal proliferator-activated receptors (PPARs) are nuclear receptors that regulate metabolic signaling pathways, cell differentiation, and immune inflammation [43]. There are three different patterns of tissue expression and functional activity. PPARa is expressed mostly in active metabolism tissues such as the liver, kidney, heart, and skeletal tissue. It has been demonstrated to lower triglycerides and enhance HDL-C in serum by regulating lipid delivery through fatty acid transport and b-oxidation [44]. PPARg regulates lipogenesis, fatty acid differentiation, and glucose homeostasis. As a sensor of insulin, PPARg reduces ectopic fat accumulation by increasing the storage of fatty acids such as TG. It can also reduce inflammation by inhibiting the production of cytokines [45]. PPARd has a ubiquitous tissue expression profile, and its function is involved in fatty acid metabolism [46].Elafibranor is a dual agonist for PPARa and PPARd that improves insulin sensitivity, glucose homeostasis, and lipid metabolism. In double-blind, placebo-controlled research, patients with NASH without cirrhosis were randomly assigned to one of three groups: elafibranor 80 mg, 120 mg, or placebo daily for 52 weeks (NCT01694849) [47]. NASH resolution was reduced with elafibranor 120 mg daily, without fibrosis worsening. It was well tolerated and did not cause weight gain. Subsequently, the effect of elafibranor 200 mg daily for 72 weeks was assessed in a Phase III trial (NCT02704403). The interim analysis, however, found no evidence of a therapeutic effect, with a response rate of 19.2% in the elafibranor arm against 14.7% in the placebo arm in NASH resolution without worsening fibrosis.
- (7)
- GLP-1 receptor agonistsGlucagon-like peptide-1 (GLP-1) is a gut-derived incretin hormone that induces weight loss and insulin sensitivity by improving insulin synthesis and release, reduces glucagon secretion and hepatic gluconeogenesis, reduces steatosis, and affects lipid metabolism by decreasing lipogenesis and increasing oxidation to metabolize fatty acids [48]. In mouse models of NASH, GLP-1 analogs have been demonstrated to lower liver enzymes, oxidative stress, and liver histopathology. Endogenous GLP-1 is degraded within minutes in vivo by the enzyme dipeptidyl peptidase-4 (DP-4), whereas liraglutide is a long-term GLP-1 receptor agonist that has been licensed for the treatment of T2DM [49].Patients with overweight and NASH were investigated in a 48-week, randomized, double-blind, placebo-controlled Phase II experiment (NCT01237119) [50]. The primary outcome resolution of definite NASH with no worsening in fibrosis was met in patients receiving liraglutide. Patients receiving liraglutide showed a significantly mean BMI decrease versus the placebo group from baseline. Unfortunately, there was no statistically significant difference in histologic response between the liraglutide and placebo groups (including hepatocyte ballooning score, steatosis, lobular inflammation, Kleiner fibrosis stage, and total NAFLD activity score), indicating that the benefits of liraglutide were possibly related to weight loss rather than liraglutide treatment. The majority of the AEs were mild to moderate in severity.A 72-week, double-blinded Phase II trial involving patients with NASH and Stage F1-3 fibrosis as confirmed by biopsy, was evaluated to compare semaglutide with placebo (NCT02970942) [51]. The primary end point of NASH remission and no worsening of fibrosis was fulfilled for all doses of semaglutide compared to placebo [52]. However, a recent study found that semaglutide failed to cure NASH with F4 fibrosis, indicating that it is difficult to reverse advanced fibrosisonly metabolic homeostasis and that combined therapy is required for advanced fibrosis and/or cirrhosis treatment.
Antibodies
3.1.2. Targeting Liver Inflammation and Cell Death
Antibodies
- (1)
- CB1 antibodyCannabinoid receptor 1 (CB1R) is a G-protein coupled receptor, whose expression is unregulated in the liver with viral hepatitis, alcoholic and nonalcoholic fatty liver disease, and cirrhosis. The inhibition of CB1R exhibited a beneficial effect in improving liver function by suppressing the activation of HSCs [54]. Nimacimab, also known as RYI-018, is an antagonist antibody, and in a multicenter, adaptive design, randomized, parallel-group Phase I trial, the safety, tolerability, and PK of repeat IV doses of RYI-018 in participants with NAFLD were examined (NCT03261739). In addition, a Phase IIb clinical trial is going to be conducted.
- (2)
- Anti-CCL24 AntibodyCCL24 is a chemokine that promotes immune cell trafficking and activation as well as profibrotic activities through the CCRs receptor. A blocking mAb that targets CCL4 and CM-101 was evaluated in a Phase 1b trial, and the results showed that CM-101 treatment decreased fibrosis and inflammatory biomarkers in serum without compromising safety or tolerability [55]. The effect and safety of CM-101 will be evaluated in NASH patients with Stage 2–3 fibrosis.
Natural Compounds
- (1)
- Berberine (BBR)Along with chemical compounds, various natural compounds have interestingly been prescribed for the treatment of liver fibrosis. Natural compounds with a diverse structure, low toxicity, and a wide range of sources have snatched up a lot of attention in recent years for their multiple-target actions in liver disease.Berberine (BBR) is a benzylisoquinoline alkaloid with various pharmacological activities, such as antiparkinson, anti-cancer, anti-diabetic, anti-obesity, anti-inflammatory, anti-viral, and cardiovascular actions. These potential biological activities of BBR were attributed due to their ability to interact with various biological targets involved in the pathogenesis of various diseases, including liver fibrosis. Based on the efficacy of HTD1801 (berberine ursodeoxycholate, ionic salt of berberine, and ursodeoxycholic acid) in animals, a Phase II randomized controlled trial of two doses of HTD1801 versus placebo in 100 patients with fatty liver disease and diabetes was conducted in 100 patients (NCT03656744) [56]; 18 weeks treatment of HTD1801 (1000 mg twice a day) reduced liver fat content assessed by MRI-PDFF (mean absolute decrease −4.8% in HTD1801 arm versus −2.0% in the placebo arm; p = 0.011) with gastrointestinal AEs.
- (2)
- SilymarinSilymarin is a lipophilic complex of two flavonoids (quercetin and taxifolin) and three flavonolignane diastereomers: silibinin, silydianin, and silychristin. A randomized, double-blind, placebo-controlled trial (NCT02006498) was conducted to assess the effect of silymarin on patients with NASH and NAFLD activity scores (NAS) of at least four in patients with NASH and NAFLD (proved by biopsy) [57]. Patients who received silymarin (700 mg daily for 48 weeks) did not have their NAS scores reduced by 30% compared to those who received a placebo. The therapeutic impact of silymarin in the treatment of NAFLD was investigated in a meta-analysis (PRISMA) of randomized control trials involving 587 people with NAFLD [58]. Silymarin was found to dramatically lower AST and ALT levels when compared to the control group, implying that it could be useful phytotherapy for NAFLD patients [59].
3.1.3. Targeting Liver Fibrosis
Antibodies
Gene Therapy
Cell Therapy
3.1.4. Combination Drug Trials
- Mechanistically complementary, such as the combination of an ACC1/2 inhibitor (PF-05221304) and a DGAT2 inhibitor (PF-06865571): In participants with NAFLD, a Phase II trial (NCT03776175) was conducted to investigate the effects of PF-05221304 alone, PF-06865571 alone, and the combination of PF-05221304 and PF-06865571 [66]. The results indicated that PF-05221304 monotherapy exhibits a great anti-steatosis effect; however, elevation in serum TG with a high dose of PF-05221304 limits the utility for those with cardio-metabolic diseases. Co-administration of PF-05221304 with PF-06865571 may be a viable approach for overcoming the limitations of PF-05221304 monotherapy and achieving better therapeutic benefits.
- Targeting different disease characteristics, such as FXR (targeting fibrosis and inflammation) and ACC (reducing fat accumulation): A combination of the ACC inhibitor GS-0976 (firsocostat) and the nonsteroidal FXR agonist GS-9674 (cilofexor) trial was observed to improve hepatic steatosis, biochemistry, and stiffness in patients with NASH (NCT02781584) [67]. However, no clinically significant reductions in total LDL or HDL were observed with any of the programs.
- Combination of small-molecule drugs tropifexor (LJN452) and cenicriviroc (CVC): A randomized, double-blind study was conducted to evaluate the safety, tolerability, and efficacy of a tropifexor (LJN452) and cenicriviroc (CVC) combination treatment in patients with NASH and liver fibrosis (NCT03517540) [68]. In addition, the results suggested that when compared to monotherapy, combination therapy is likely to provide extra benefits.
- Small-molecule drug medications combined with macromolecular drugs: In patients with NASH, the FXR agonist cilofexor (GS-9674), the acetyl-coenzyme A carboxylase inhibitor firsocostat (GS-0976), and the GLP1 receptor agonist semaglutide were evaluated (NCT03987074) [69]. Semaglutide plus firsocostat and/or cilofexor was generally well tolerated in patients with mild-to-moderate fibrosis owing to NASH. Treatment resulted in additional improvements in liver steatosis and biochemistry compared to semaglutide alone in exploratory efficacy studies.
- Dual-target macromolecular drugs: LY3298176 is a novel dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that has been developed for the treatment of type 2 diabetes [70]. GLP1 receptor agonists and dual GLP1-GIP agonists have been reported to be associated with significant weight loss approaching 10% or more. In a Phase II randomized, double-blind trial (NCT03131687), LY3298176 was found to have significantly greater efficacy than GLP1R agonist in terms of glucose management and weight loss, with an acceptable safety and tolerability profile. According to the most recent data, individuals taking trizepatide 15 mg once a week lost a significant amount of weight (−22.5%), demonstrating its efficacy in the treatment of NASH with fatty liver. At 26 weeks, the 10 mg and 15 mg tirzmepatide groups showed significant reductions in ALT, AST, K-18, and Pro-C3, whereas adiponectin increased significantly. In order to compare the safety and efficacy of tirzmepatide vs. placebo in individuals with NASH, 196 people were recruited (SYNERGY-NASH, NCT04166773). Mounjaro (tirzepatide), developed by Eli Lilly, has been granted FDA and EMAs approval for use in combination with diet and exercise to improve glycemic control in adults with type 2 diabetes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Categories | Agent | Phase of Clinical Trial | Ref. |
|---|---|---|---|---|
| Targeting liver lipid metabolism and oxidative stress | Molecule compounds | ACC inhibitors Firsocostat PF-05221304 | Phase II Phase II | [27,70,71] |
| SCD1 inhibitors Aramchol | Phase III | [28] | ||
| FGF agonists Aldafermin Pegbelfermin Efruxifermin | Phase II Phase II Phase II | [29,30,31,32] | ||
| THR beta agonists Resmetirom K2809-201 | Phase III Phase II | [35,36,37] | ||
| FXR agonists Obeticholic acid Tropifexor Cilofexor | Phase III Phase II Phase II | [38,39,40,42,72] | ||
| PPAR agonists Elafibranor Lanifibranor Saroglitazar Seladelpar | Phase III Phase II Phase II Phase II | [47,73,74,75] | ||
| ASK1 inhibitors Selonsertib | Phase III | [76] | ||
| GLP-1 receptor agonists Liraglutide Semaglutide Trizepatide | Phase II Phase II Phase II | [50,51,52,77] | ||
| SGLT inhibitors Ipragliflozin Dapagliglozin | Phase II Phase II | [78] | ||
| Antibodies | FGFR1c/KLB | Phase II | [53] | |
| Targeting liver inflammation and cell death | Molecule compounds | CCR2/CCR5 inhibitors Cenicriviroc | Phase III | [79] |
| Mitochondrial pyruvate carrier MSDC-0602K | Phase II | [80] | ||
| Antibodies | CB1 antibody Nimacimab | Phase I | [54] | |
| Anti-IL-17 antibody Secukinumab | Phase I | [81] | ||
| Anti-CCL24 antibody CM-101 | Phase II | [55] | ||
| Natural compounds | Berberine (BBR) | Phase II | [56] | |
| Silymarin | Phase II | [57] | ||
| Resveratrol (RES) | Phase II | [82] | ||
| Targeting liver fibrosis | Molecule compounds | Galectin 3 inhibitors GR-MD-02 | Phase III | [83] |
| ARB inhibitors Losartan | Phase III | [84,85] | ||
| Tyrosine kinase inhibitors Sorafenib | Phase II | [86,87] | ||
| Antibodies | LOXL2 inhibitors | Phase II | [88] | |
| Integrin aV inhibitors 3G9 | Phase II | [89] | ||
| CD105 inhibitors TRC105 | Phase II | [60] | ||
| Gene therapy | siHSP47 | Phase II | [62] |
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular Carcinoma |
| HSC | Hepatic Stellate Cells |
| NASH | Nonalcoholic Steatohepatitis |
| IR | Insulin Resistance |
| FFA | Free Fatty Acids |
| MMPs | Metalloproteinases |
| ECM | Extracellular Matrix Protein |
| LSECs | Liver Sinusoidal Endothelial Cells |
| TIMPs | Tissue Inhibitors of MMPs |
| LFC | Liver Fat Content |
| FGF | Fibroblast growth factor |
| HFF | Hepatic Fat Fraction |
| THR | Thyroid Hormone Receptor |
| FXR | Farnesoid X Receptor |
| VLDL | Very Low-density Lipoprotein |
| HDL | High-density Lipoprotein |
| PPARs | Peroxisomal Proliferator-activated Receptors |
| GLP-1 | Glucagon-like Peptide-1 |
| GIP | Gastric Inhibitory Polypeptide |
| CCR2 | C-C chemokine receptor 2 |
| MPC | Mitochondrial Pyruvate Carrier |
| ALD | Alcoholic Liver Disease |
| HVPG | Hepatic Venous Pressure Gradient |
| HSP47 | Heat Shock Protein 47 |
| siRNA | Small Interfering RNA |
| ACE-1 | Angiotensin-I Converting Enzyme 1 |
| AT-II | Angiotensin-II |
| RAS | Renin-angiotensin System |
| ADCC | Antibody-dependent Cellular Cytotoxicity |
| PDGF | Platelet-derived Growth Factor |
| VEGF | Vascular Endothelial Growth Factor |
| FGF | Fibroblast Growth Factor |
| EGF | Epidermal Growth Factor |
| PVP | Portal Vein Pressure |
| MCD | Methionine and Choline-deficient Diet |
| ROS | Reactive Oxygen Species |
| HFD | High-Fat Diet |
| CYP2E1 | Cytochrome P450 2E1 |
| HA | Hyaluronic Acid |
| Bcl-2 | B-cell Lymphoma-2 |
| HFHS | High-Fat High-Sucrose |
| ALP | Alkaline Phosphatase |
| a-SMA | alpha-Smooth Muscle Actin |
| TGF-b1 | Transforming Growth Factor-b 1 |
| CDAA | Choline-deficient-amino-acid–defined |
| CPT-1 | Carnitine Palmitoyl Transferase-1 |
| ASGPR | Asialoglycoprotein Receptor |
| Chol | Cholesterol |
| IGF2R | Insulin-like Growth Factor 2 Receptor |
| CD44 | Cluster of Differentiation 44 |
| RBP | Retinol-binding Protein |
| TRAIL | TNF-related Apoptosis-inducing Ligand |
| NPs | Nanoparticles |
| cRGD | cyclic RGD |
| LNPs | Lipid Nanoparticles |
References
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Li, Y. Sorafenib-Loaded Ligand-Functionalized Polymer-Lipid Hybrid Nanoparticles for Enhanced Therapeutic Effect Against Liver Cancer. J. Nanosci. Nanotechnol. 2019, 19, 6866–6871. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7, F1000 Faculty Rev-921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Kurniawan, D.W.; Jajoriya, A.K.; Dhawan, G.; Mishra, D.; Argemi, J.; Bataller, R.; Storm, G.; Mishra, D.P.; Prakash, J.; Bansal, R. Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis. J. Control. Release 2018, 288, 227–238. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Ji, D.; Wang, Q.; Zhao, Q.; Tong, H.; Yu, M.; Wang, M.; Lu, T.; Jiang, C. Co-delivery of miR-29b and germacrone based on cyclic RGD-modified nanoparticles for liver fibrosis therapy. J. Nanobiotechnol. 2020, 18, 86. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.G.; Moon, M.J.; Kim, J.H.; Lee, J.H.; Jeong, Y.Y. Effectiveness of Losartan-Loaded Hyaluronic Acid (HA) Micelles for the Reduction of Advanced Hepatic Fibrosis in C3H/HeN Mice Model. PLoS ONE 2015, 10, e0145512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.; Nakagawa, S.; Higashi, T.; Vincek, A.; Venkatesh, A.; Ruiz de Galarreta, M.; Koh, A.P.; Goossens, N.; Hirschfield, H.; Bian, C.B. Cell type-specific pharmacological kinase inhibition for cancer chemoprevention. Nanomedicine 2018, 14, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Hunt, N.J.; Lockwood, G.P.; Le Couteur, F.H.; McCourt, P.; Singla, N.; Kang, S.; Burgess, A.; Kuncic, Z.; Le Couteur, D.G.; Cogger, V.C. Rapid Intestinal Uptake and Targeted Delivery to the Liver Endothelium Using Orally Administered Silver Sulfide Quantum Dots. ACS Nano 2020, 14, 1492–1507. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; IT-LIVER Consortium. TGF-b signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-b activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68, 28–43. [Google Scholar] [CrossRef]
- Fan, W.; Liu, T.; Chen, W.; Hammad, S.; Longerich, T.; Hausser, I.; Fu, Y.; Li, N.; He, Y.; Liu, C.; et al. ECM1 Prevents Activation of Transforming Growth Factor b, Hepatic Stellate Cells, and Fibrogenesis in Mice. Gastroenterology 2019, 157, 352–1367. [Google Scholar] [CrossRef]
- Duspara, K.; Bojanic, K.; Pejic, J.I.; Kuna, L.; Kolaric, T.O.; Nincevic, V.; Smolic, R.; Vcev, A.; Glasnovic, M.; Curcic, I.B.; et al. Targeting the Wnt Signaling Pathway in Liver Fibrosis for Drug Options: An Update. J. Clin. Transl. Hepatol. 2021, 9, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Akcora, B.Ö.; Storm, G.; Bansal, R. Inhibition of canonical WNT signaling pathway by b-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Im, E.; Lee, H.J.; Sim, D.Y.; Lee, J.H.; Jung, J.H.; Park, J.E.; Shim, B.S.; Kim, S.H. Apoptotic and antihepatofibrotic effect of honokiol via activation of GSK3b and suppression of Wnt/b-catenin pathway in hepatic stellate cells. Phytother. Res. 2021, 35, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef]
- Li, Y.; Pu, S.; Liu, Q.; Li, R.; Zhang, J.; Wu, T.; Chen, L.; Li, H.; Yang, X.; Zou, M.; et al. An integrin-based nanoparticle that targets activated hepatic stellate cells and alleviates liver fibrosis. J. Control. Release 2019, 303, 77–90. [Google Scholar] [CrossRef]
- Schuppan, D.; Myneni, S.; Surabattula, R. Liquid biomarkers for fibrotic NASH—Progress in a complex field. J. Hepatol. 2022, 76, 5–7. [Google Scholar] [CrossRef]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463–1473. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled Phase 2b trial. Nat. Med. 2021, 27, 1825–1835. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients with Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Ling, L.; Beuers, U.; DePaoli, A.M.; Lieu, H.D.; Harrison, S.A.; Hirschfield, G.M. Potent suppression of hydrophobic bile acids by aldafermin, an FGF19 analogue, across metabolic and cholestatic liver diseases. JHEP Rep. 2021, 3, 100255. [Google Scholar] [CrossRef]
- Verzijl, C.R.C.; Van De Peppel, I.P.; Struik, D.; Jonker, J.W. Pegbelfermin (BMS-986036): An investigational PEGylated fibroblast growth factor 21 analogue for the treatment of nonalcoholic steatohepatitis. Expert Opin. Investig. Drugs 2020, 29, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, Phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled, Phase 2a trial. Nat. Med. 2021, 27, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, W.; Joo, S.K.; Bae, J.M.; Kim, J.H.; Ahmed, A. Subclinical Hypothyroidism and Low-Normal Thyroid Function Are Associated with Nonalcoholic Steatohepatitis and Fibrosis. Clin. Gastroenterol. Hepatol. 2018, 16, 123–131. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Carr, R.M.; Reid, A.E. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr. Atheroscler. Rep. 2015, 17, 500. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2020, 72, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Tully, D.C.; Rucker, P.V.; Chianelli, D.; Williams, J.; Vidal, A.; Alper, P.B.; Mutnick, D.; Bursulaya, B.; Schmeits, J.; Wu, X.; et al. Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J. Med. Chem. 2017, 24, 9960–9973. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients with Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, G.A.; Prins, J.B. Peroxisomal fatty acid metabolism, peroxisomal proliferator-activated receptors and non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2004, 19, 1335–1337. [Google Scholar] [CrossRef]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Montagner, A.; Tan, N.S.; Wahli, W. Insights into the Role of PPARb/d in NAFLD. Int. J. Mol. Sci. 2018, 19, 1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-a and -d, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159, reprinted in Gastroenterology 2017, 152, 2084. [Google Scholar] [CrossRef] [Green Version]
- Htike, Z.Z.; Zaccardi, F.; Papamargaritis, D.; Webb, D.R.; Khunti, K.; Davies, M.J. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: A systematic review and mixed-treatment comparison analysis. Diabetes Obes. Metab. 2017, 19, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor a/d agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; NN9931-4296 Investigators. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Calanna, S.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.; Sejling, A.S.; Newsome, P.N. Semaglutide for the treatment of non-alcoholic steatohepatitis: Trial design and comparison of non-invasive biomarkers. Contemp. Clin. Trials 2020, 97, 106174. [Google Scholar] [CrossRef] [PubMed]
- Baruch, A.; Wong, C.; Chinn, L.W.; Vaze, A.; Sonoda, J.; Gelzleichter, T.; Chen, S.; Lewin-Koh, N.; Morrow, L.; Dheerendra, S.; et al. Antibody-mediated activation of the FGFR1/Klothobeta complex corrects metabolic dysfunction and alters food preference in obese humans. Proc. Natl. Acad. Sci. USA 2020, 117, 28992–29000. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Gautam, S.; Aseer, K.R.; Kim, J.; Chandrasekaran, P.; Mazucanti, C.H.; Ghosh, P.; O’Connell, J.F.; Doyle, M.E.; Appleton, A.; et al. Hepatocyte cannabinoid 1 receptor nullification alleviates toxin-induced liver damage via NF-kB signaling. Cell Death Dis. 2020, 11, 1044. [Google Scholar] [CrossRef]
- Segal-Salto, M.; Barashi, N.; Katav, A.; Edelshtein, V.; Aharon, A.; Hashmueli, S.; George, J.; Maor, Y.; Pinzani, M.; Haberman, D.; et al. A blocking monoclonal antibody to CCL24 alleviates liver fibrosis and inflammation in experimental models of liver damage. JHEP Rep. 2020, 2, 100064. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Gunn, N.; Neff, G.W.; Kohli, A.; Liu, L.; Flyer, A.; Goldkind, L.; Di Bisceglie, A.M. A phase 2, proof of concept, randomised controlled trial of berberine ursodeoxycholate in patients with presumed non-alcoholic steatohepatitis and type 2 diabetes. Nat. Commun. 2021, 12, 5503. [Google Scholar] [CrossRef]
- Kheong, C.W.; Mustapha, N.R.N.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 1940–1949. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Fan, Y.; Yan, Q.; Fan, X.; Wu, B.; Han, Y.; Zhang, Y.; Chen, Y.; Zhang, H.; Niu, J. The therapeutic effect of silymarin in the treatment of nonalcoholic fatty disease: A meta-analysis (PRISMA) of randomized control trials. Medicine 2017, 96, e9061. [Google Scholar] [CrossRef]
- Kalopitas, G.; Antza, C.; Doundoulakis, I.; Siargkas, A.; Kouroumalis, E.; Germanidis, G.; Samara, M.; Chourdakis, M. Impact of Silymarin in individuals with nonalcoholic fatty liver disease: A systematic review and meta-analysis. Nutrition 2021, 83, 111092. [Google Scholar] [CrossRef]
- Wu, H.W.; Sheard, M.A.; Malvar, J.; Fernandez, G.E.; DeClerck, Y.A.; Blavier, L.; Shimada, H.; Theuer, C.P.; Sposto, R.; Seeger, R.C. Anti-CD105 Antibody Eliminates Tumor Microenvironment Cells and Enhances Anti-GD2 Antibody Immunotherapy of Neuroblastoma with Activated Natural Killer Cells. Clin. Cancer Res. 2019, 25, 4761–4774. [Google Scholar] [CrossRef]
- Nagata, K. Hsp47: A collagen-specific molecular chaperone. Trends Biochem. Sci. 1996, 21, 22–26. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Shevell, D.E.; Tirucherai, G.S.; Du, S.; Chen, W.; Kavita, U.; Coste, A.; Poordad, F.; Karsdal, M.; Nielsen, M.; et al. BMS-986263 in patients with advanced hepatic fibrosis: 36-week results from a randomized, placebo-controlled phase 2 trial. Hepatology 2022, 75, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Ji, C.; Lu, L. Mesenchymal stem cell therapy for liver fibrosis/cirrhosis. Ann. Transl. Med. 2022, 8, 562. [Google Scholar] [CrossRef]
- Liu, Q.; Lv, C.; Huang, Q.; Zhao, L.; Sun, X.; Ning, D.; Liu, J.; Jiang, Y.; Jin, S. ECM1 modified HF-MSCs targeting HSC attenuate liver cirrhosis by inhibiting the TGF-b/Smad signaling pathway. Cell Death Discov. 2022, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Calle, R.A.; Amin, N.B.; Carvajal-Gonzalez, S.; Ross, T.T.; Bergman, A.; Aggarwal, S.; Crowley, C.; Rinaldi, A.; Mancuso, J.; Aggarwal, N.; et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: Two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 2021, 27, 1836–1848. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Noureddin, M.; Kowdley, K.V.; Kohli, A.; Sheikh, A.; Neff, G.; Bhandari, B.R.; Gunn, N.; Caldwell, S.H.; Goodman, Z.; et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 2021, 73, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.; Seyedkazemi, S.; Francque, S.; Sanyal, A.; Rinella, M.; Charlton, M.; Loomba, R.; Ratziu, V.; Kochuparampil, J.; Fischer, L.; et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp. Clin. Trials 2020, 88, 105889. [Google Scholar] [CrossRef] [Green Version]
- Alkhouri, N.; Herring, R.; Kabler, H.; Kayali, Z.; Hassanein, T.; Kohli, A.; Huss, R.S.; Zhu, Y.; Billin, A.N.; Damgaard, L.H.; et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J. Hepatol. 2022, 77, 607–618. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Bergman, A.; Carvajal-Gonzalez, S.; Tarabar, S.; Saxena, A.R.; Esler, W.P.; Amin, N.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of a Liver-Targeting Acetyl-CoA Carboxylase Inhibitor (PF-05221304): A Three-Part Randomized Phase 1 Study. Clin. Pharmacol. Drug Dev. 2020, 9, 514–526. [Google Scholar] [CrossRef]
- Huard, K.; Smith, A.C.; Cappon, G.; Dow, R.L.; Edmonds, D.J.; El-Kattan, A.; Esler, W.P.; Fernando, D.; Griffith, D.A.; Kalgutkar, A.S.; et al. Optimizing the Benefit/Risk of Acetyl-CoA Carboxylase Inhibitors through Liver Targeting. J. Med. Chem. 2020, 63, 10879–10896. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non-Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andres-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernandez-Iglesias, A.; Broqua, P.; Junien, J.L.; Wettstein, G.; Bosch, J.; et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Ranvir, R.; Kadam, S.; Patel, H.; Swain, P.; et al. Dual PPARa/g agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018, 38, 1084–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559, reprinted in Hepatology 2018, 67, 2063. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients with Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef]
- Honda, Y.; Imajo, K.; Kato, T.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; Mawatari, H.; Fujita, K.; Yoneda, M.; et al. The Selective SGLT2 Inhibitor Ipragliflozin Has a Therapeutic Effect on Nonalcoholic Steatohepatitis in Mice. PLoS ONE 2016, 11, e0146337. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef] [Green Version]
- Colca, J.R.; McDonald, W.G.; Adams, W.J. MSDC-0602K, a metabolic modulator directed at the core pathology of non-alcoholic steatohepatitis. Expert Opin. Investig. Drugs 2018, 27, 631–636. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, B.T.; Rodriguez-Bies, E.; Talero, E.; Gamero-Estevez, E.; Motilva, V.; Navas, P.; Lopez-Lluch, G. Anti-inflammatory effect of resveratrol in old mice liver. Exp. Gerontol. 2015, 64, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Noguchi, R.; Ikenaka, Y.; Kitade, M.; Kaji, K.; Tsujimoto, T.; Uemura, M.; Fukui, H. Renin-angiotensin system inhibitors as therapeutic alternatives in the treatment of chronic liver diseases. Curr. Med. Chem. 2007, 14, 2749–2754. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bataller, R.; Dulyx, J.; Coffman, T.M.; Gines, P.; Rippe, R.A.; Brenner, D.A. Attenuated hepatic inflammation and fibrosis in angiotensin type 1a receptor deficient mice. J. Hepatol. 2005, 43, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Yokohama, S.; Yoneda, M.; Haneda, M.; Okamoto, S.; Okada, M.; Aso, K.; Hasegawa, T.; Tokusashi, Y.; Miyokawa, N.; Nakamura, K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 2004, 40, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Huang, Z.; Lin, T.; Liu, S.; Chang, H.; Yan, Z.; Zhang, H.; Liu, C. New Insight into the Anti-liver Fibrosis Effect of Multitargeted Tyrosine Kinase Inhibitors: From Molecular Target to Clinical Trials. Front. Pharmacol. 2016, 6, 300. [Google Scholar] [CrossRef] [Green Version]
- Mejias, M.; Garcia-Pras, E.; Tiani, C.; Miquel, R.; Bosch, J.; Fernandez, M. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology 2009, 49, 1245–1256. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients with Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Zhang, Y.; Sun, B. The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application. Int. J. Mol. Sci. 2022, 23, 12572. https://doi.org/10.3390/ijms232012572
Zhang D, Zhang Y, Sun B. The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application. International Journal of Molecular Sciences. 2022; 23(20):12572. https://doi.org/10.3390/ijms232012572
Chicago/Turabian StyleZhang, Danyan, Yaguang Zhang, and Bing Sun. 2022. "The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application" International Journal of Molecular Sciences 23, no. 20: 12572. https://doi.org/10.3390/ijms232012572
APA StyleZhang, D., Zhang, Y., & Sun, B. (2022). The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application. International Journal of Molecular Sciences, 23(20), 12572. https://doi.org/10.3390/ijms232012572