

Rationale for Combining the BCL2 Inhibitor Venetoclax with the PI3K Inhibitor Bimiralisib in the Treatment of IDH2- and FLT3-Mutated Acute Myeloid Leukemia



Abstract
:1. Introduction
2. Results
2.1. Variable Susceptibility of AML Cell Lines to Venetoclax and Various Targeted Therapies
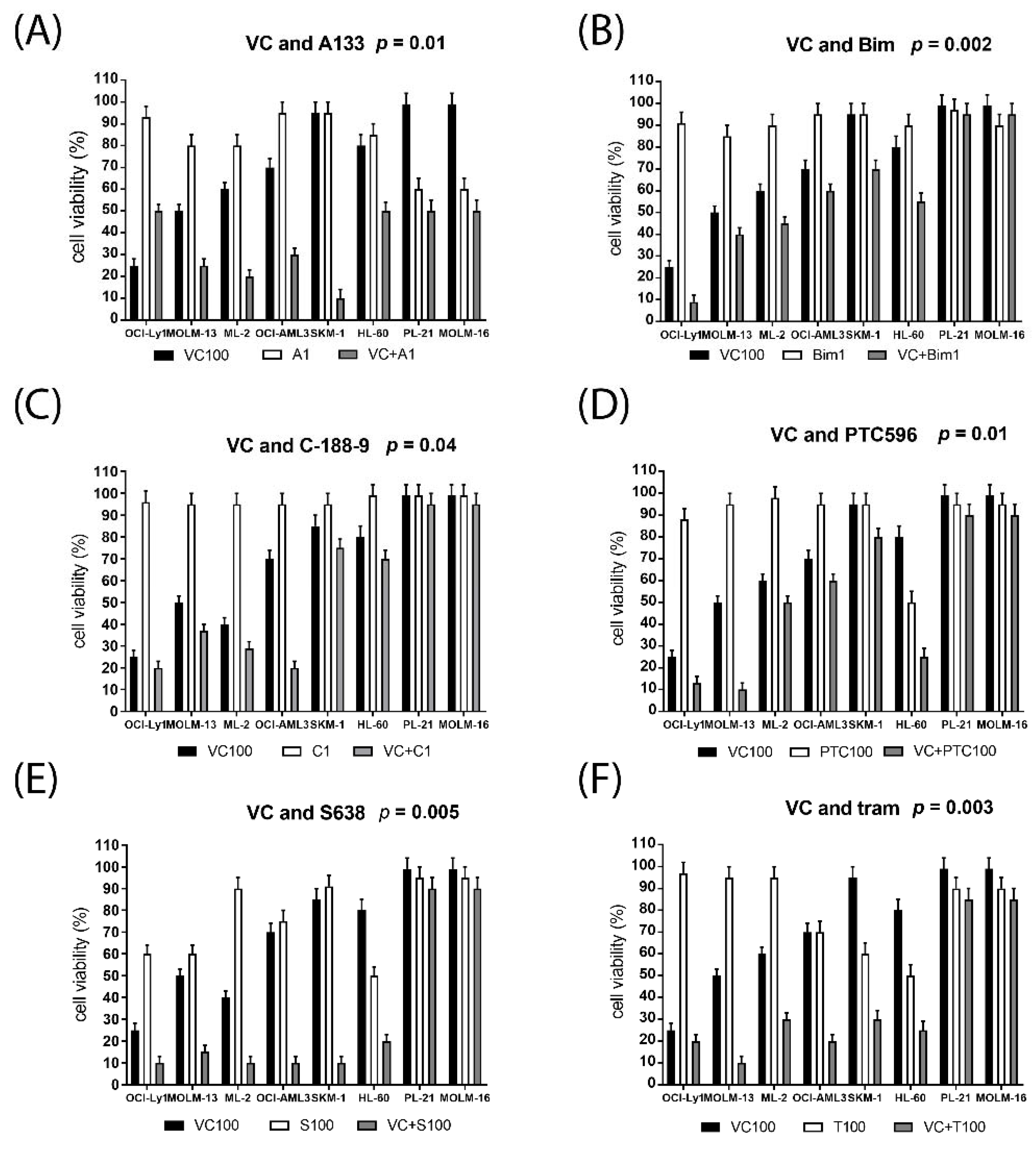
2.2. Synergistic Effects on Cell Viability in AML Cell Lines Treated with Venetoclax Combinations
2.3. Altered Susceptibility to Targeted Therapies in AML Cells Grown in the Presence of Bone Marrow Stroma
2.4. Venetoclax Combination Treatment Induces Cell Cycle Arrest, Apoptosis and Cell Death
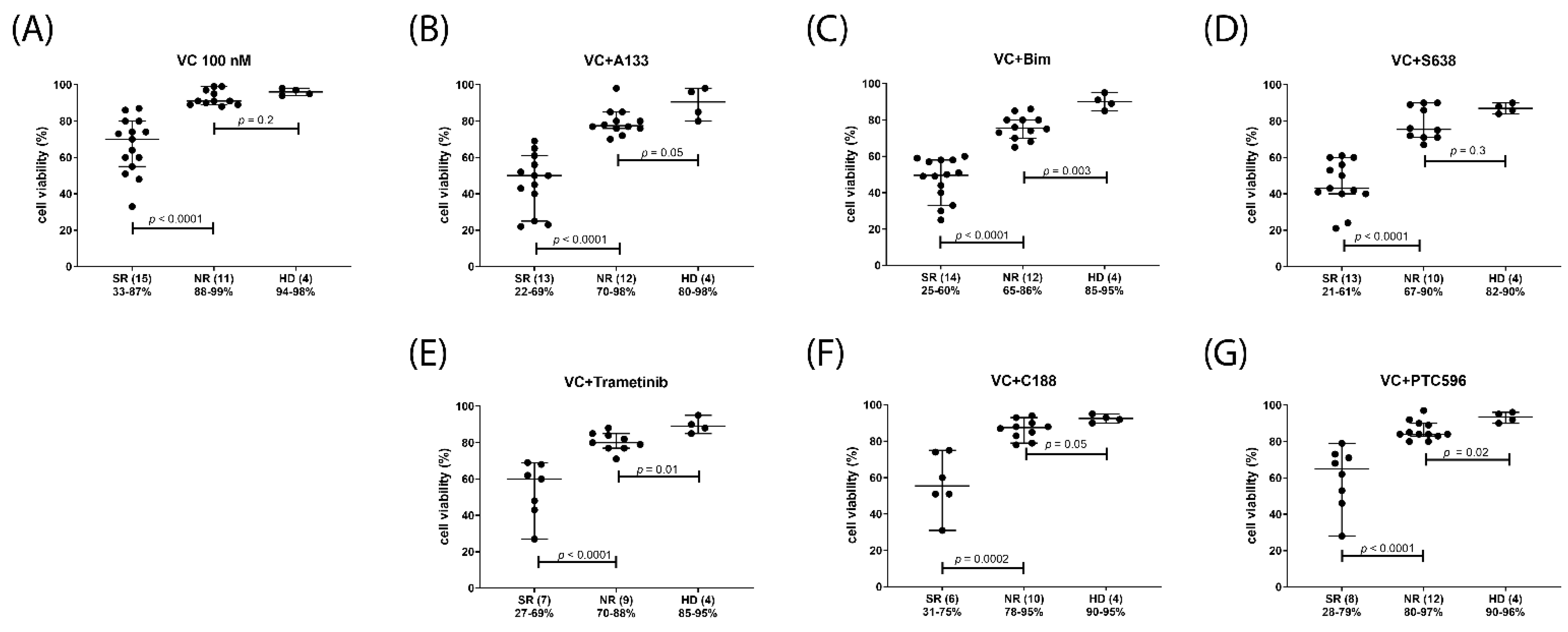
2.5. Venetoclax Combination Treatments with Differential Efficacy in Subsets of AML Patients
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Cell Lines and Cell Culture
4.3. Cytotoxicity Assays
4.4. Imaging Cytometry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fleischmann, M.; Schnetzke, U.; Hochhaus, A.; Scholl, S. Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives. Cancers 2021, 13, 5722. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Ganser, A. Treatment of Relapsed Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2020, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular Patterns of Response and Treatment Failure after Frontline Venetoclax Combinations in Older Patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Stahl, M.; Menghrajani, K.; Derkach, A.; Chan, A.; Xiao, W.; Glass, J.; King, A.C.; Daniyan, A.F.; Famulare, C.; Cuello, B.M.; et al. Clinical and Molecular Predictors of Response and Survival Following Venetoclax Therapy in Relapsed/Refractory AML. Blood Adv. 2021, 5, 1552–1564. [Google Scholar] [CrossRef]
- Salah, H.T.; DiNardo, C.D.; Konopleva, M.; Khoury, J.D. Potential Biomarkers for Treatment Response to the BCL-2 Inhibitor Venetoclax: State of the Art and Future Directions. Cancers 2021, 13, 2974. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.; Tremblay, D.; Dougherty, M.; Czaplinska, T.; Sanchez, G.; Brady, C.; Kremyanskaya, M.; Bar-Natan, M.; Keyzner, A.; Marcellino, B.K.; et al. Safety and Efficacy: Clinical Experience of Venetoclax in Combination With Hypomethylating Agents in Both Newly Diagnosed and Relapsed/Refractory Advanced Myeloid Malignancies. Hemasphere 2021, 5, e549. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.; Tang, K.; Al-Abri, M.; Hall, V.; Tremblay-Lemay, R.; Rashedi, I.; Tsui, H.; Chan, S.M. RUNX1 Mutations Correlate with Response to Venetoclax Combination Therapies in Relapsed/Refractory Acute Myeloid Leukemia. Leuk. Res. 2021, 111, 106735. [Google Scholar] [CrossRef] [PubMed]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic Biomarkers Of Sensitivity and Resistance to Venetoclax Monotherapy in Patients With Relapsed Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, E202–E205. [Google Scholar] [CrossRef] [Green Version]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Ewald, L.; Dittmann, J.; Vogler, M.; Fulda, S. Side-by-Side Comparison of BH3-Mimetics Identifies MCL-1 as a Key Therapeutic Target in AML. Cell Death Dis. 2019, 10, 917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, V.M.; Dietz, A.; Henz, K.; Bruecher, D.; Jackson, R.; Kowald, L.; van Wijk, S.J.L.; Jayne, S.; Macip, S.; Fulda, S.; et al. Specific Interactions of BCL-2 Family Proteins Mediate Sensitivity to BH3-Mimetics in Diffuse Large B-Cell Lymphoma. Haematologica 2020, 105, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 Inhibitor S63845 Is Tolerable and Effective in Diverse Cancer Models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Seipel, K.; Schmitter, K.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy Consisting of MCL1- and MEK-Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipel, K.; Kopp, B.; Bacher, U.; Pabst, T. BMI1-Inhibitor PTC596 in Combination with MCL1 Inhibitor S63845 or MEK Inhibitor Trametinib in the Treatment of Acute Leukemia. Cancers 2021, 13, 581. [Google Scholar] [CrossRef] [PubMed]
- Kevlicius, L.; Cepulyte, R.; Vasilevska, D.; Griskevicius, L.; Zucenka, A. Venetoclax-Based Regimens in Combination with Trametinib for RAS-Mutated Relapsed or Refractory Myeloid Malignancies. Bone Marrow Transpl. 2022, 57, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Seipel, K.; Graber, C.; Flückiger, L.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 8092. [Google Scholar] [CrossRef]
- Han, L.; Zhang, Q.; Dail, M.; Shi, C.; Cavazos, A.; Ruvolo, V.R.; Zhao, Y.; Kim, E.; Rahmani, M.; Mak, D.H.; et al. Concomitant Targeting of BCL2 with Venetoclax and MAPK Signaling with Cobimetinib in Acute Myeloid Leukemia Models. Haematologica 2020, 105, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, F.; Stirnweiß, F.U.; Niedermayer, A.; Enzenmüller, S.; Hörl, R.L.; Münch, V.; Köhrer, S.; Debatin, K.-M.; Meyer, L.H. Synergistic Activity of Combined Inhibition of Anti-Apoptotic Molecules in B-Cell Precursor ALL. Leukemia 2022, 36, 901–912. [Google Scholar] [CrossRef]
- Bewarder, M.; Stilgenbauer, S.; Thurner, L.; Kaddu-Mulindwa, D. Current Treatment Options in CLL. Cancers 2021, 13, 2468. [Google Scholar] [CrossRef] [PubMed]
- Wierda, W.G.; Tambaro, F.P. How I Manage CLL with Venetoclax-Based Treatments. Blood 2020, 135, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, D.; Nogami, A.; Okada, K.; Akiyama, H.; Umezawa, Y.; Miura, O. FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating MTORC1 and EIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers 2019, 11, 1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-MTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [Green Version]
- Darici, S.; Alkhaldi, H.; Horne, G.; Jørgensen, H.G.; Marmiroli, S.; Huang, X. Targeting PI3K/Akt/MTOR in AML: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. [Google Scholar] [CrossRef]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaufils, F.; Cmiljanovic, N.; Cmiljanovic, V.; Bohnacker, T.; Melone, A.; Marone, R.; Jackson, E.; Zhang, X.; Sele, A.; Borsari, C.; et al. 5-(4,6-Dimorpholino-1,3,5-Triazin-2-Yl)-4-(Trifluoromethyl)Pyridin-2-Amine (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class I PI3K/MTOR Inhibitor as Clinical Candidate in Oncology. J. Med. Chem. 2017, 60, 7524–7538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/MTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.P.; Eyre, T.A.; Schmitz-Rohmer, D.; Townsend, W.; Popat, R.; Giulino-Roth, L.; Fields, P.A.; Krasniqi, F.; Soussain, C.; Stathis, A.; et al. A Phase II Study to Assess the Safety and Efficacy of the Dual MTORC1/2 and PI3K Inhibitor Bimiralisib (PQR309) in Relapsed, Refractory Lymphoma. Hemasphere 2021, 5, e656. [Google Scholar] [CrossRef]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The Novel BMI-1 Inhibitor PTC596 Downregulates MCL-1 and Induces P53-Independent Mitochondrial Apoptosis in Acute Myeloid Leukemia Progenitor Cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Jin, Q.; Fu, Q.; You, P.; Jiang, X.; Yuan, Q.; Huang, H. Induction of Multidrug Resistance of Acute Myeloid Leukemia Cells by Cocultured Stromal Cells via Upregulation of the PI3K/Akt Signaling Pathway. Oncol. Res. 2016, 24, 215–223. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Brown, N.; Xyrafas, A.; Bize, V.; Hawle, H.; Berardi, S.; Cmiljanović, N.; Cmiljanović, V.; Stumm, M.; Dimitrijević, S.; et al. First-in Human, Phase 1, Dose-Escalation Pharmacokinetic and Pharmacodynamic Study of the Oral Dual PI3K and MTORC1/2 Inhibitor PQR309 in Patients with Advanced Solid Tumors (SAKK 67/13). Eur. J. Cancer 2018, 96, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Doherty, G.A.; Judd, A.S.; Tao, Z.-F.; Hansen, T.M.; Frey, R.R.; Song, X.; Bruncko, M.; Kunzer, A.R.; Wang, X.; et al. Discovery of A-1331852, a First-in-Class, Potent, and Orally-Bioavailable BCL-XL Inhibitor. ACS Med. Chem. Lett. 2020, 11, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; O’Mara, E.; Laskin, O.L.; Gao, L.; Baird, J.D.; Spiegel, R.J.; Kaushik, D.; Weetall, M.; Colacino, J.; O’Keefe, K.; et al. Pharmacokinetics and Safety of PTC596, a Novel Tubulin-Binding Agent, in Subjects With Advanced Solid Tumors. Clin. Pharmacol. Drug Dev. 2021, 10, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M.; Dutriaux, C.; Bosco-Lévy, P.; Prey, S.; Pham-Ledard, A.; Dousset, L.; Gérard, E.; Bouchet, S.; Canal-Raffin, M.; Titier, K.; et al. Trough Dabrafenib Plasma Concentrations Can Predict Occurrence of Adverse Events Requiring Dose Reduction in Metastatic Melanoma. Clin. Chim. Acta 2017, 472, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Eckols, T.K.; Xu, X.; Kasembeli, M.M.; Chen, Y.; Adachi, M.; Song, Y.; Mo, Q.; Lai, S.Y.; Tweardy, D.J. Small-Molecule Inhibition of STAT3 in Radioresistant Head and Neck Squamous Cell Carcinoma. Oncotarget 2016, 7, 26307–26330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redell, M.S.; Ruiz, M.J.; Alonzo, T.A.; Gerbing, R.B.; Tweardy, D.J. Stat3 Signaling in Acute Myeloid Leukemia: Ligand-Dependent and -Independent Activation and Induction of Apoptosis by a Novel Small-Molecule Stat3 Inhibitor. Blood 2011, 117, 5701–5709. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; White, S.M.; Schaefer, T.S.; Ball, E.D.; Dyer, K.F.; Tweardy, D.J. Granulocyte Colony-Stimulating Factor Activation of Stat3α and Stat3β in Immature Normal and Leukemic Human Myeloid Cells. Blood 1996, 88, 2442–2449. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.; Wang, B.; You, R.; Wang, X.; Liu, J.; Zhan, W.; Chen, P.; Qin, T.; Zhang, X.; Huang, H. Stromal Cells Promote Chemoresistance of Acute Myeloid Leukemia Cells via Activation of the IL-6/STAT3/OXPHOS Axis. Ann. Transl. Med. 2020, 8, 1346. [Google Scholar] [CrossRef]
- Guan, D.; Qing, W.; Ma, C.; Zhang, Z.; Wei, H.; Wu, G. Bone Marrow Stromal-Cell Line HS-5 Affects Apoptosis of Acute Myeloid Leukemia Cells HL-60 through GLI1 Activation. Biomed Res 2018, 29, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Adamo, A.; Delfino, P.; Gatti, A.; Bonato, A.; Takam Kamga, P.; Bazzoni, R.; Ugel, S.; Mercuri, A.; Caligola, S.; Krampera, M. HS-5 and HS-27A Stromal Cell Lines to Study Bone Marrow Mesenchymal Stromal Cell-Mediated Support to Cancer Development. Front. Cell Dev. Biol. 2020, 8, 584232. [Google Scholar] [CrossRef] [PubMed]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolandi, S.M.; Pakjoo, M.; Beigi, P.; Kiani, M.; Allahgholipour, A.; Goudarzi, N.; Khorashad, J.S.; Eiring, A.M. A Role for the Bone Marrow Microenvironment in Drug Resistance of Acute Myeloid Leukemia. Cells 2021, 10, 2833. [Google Scholar] [CrossRef]
- Hormi, M.; Birsen, R.; Belhadj, M.; Huynh, T.; Cantero Aguilar, L.; Grignano, E.; Haddaoui, L.; Guillonneau, F.; Mayeux, P.; Hunault, M.; et al. Pairing MCL-1 Inhibition with Venetoclax Improves Therapeutic Efficiency of BH3-Mimetics in AML. Eur. J. Haematol. 2020, 105, 588–596. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Warmoes, M.; Lorenzi, P.L.; Mak, D.; Ruvolo, V.; Tan, L.; Cidado, J.; Drew, L.; et al. Targeting MCL-1 Dysregulates Cell Metabolism and Leukemia-Stroma Interactions and Resensitizes Acute Myeloid Leukemia to BCL-2 Inhibition. Haematologica 2022, 107, 58–76. [Google Scholar] [CrossRef]
- Zhang, H.; Nakauchi, Y.; Köhnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated Analysis of Patient Samples Identifies Biomarkers for Venetoclax Efficacy and Combination Strategies in Acute Myeloid Leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef]
- Rahmani, N.E.; Ramachandra, N.; Sahu, S.; Gitego, N.; Lopez, A.; Pradhan, K.; Bhagat, T.D.; Gordon-Mitchell, S.; Pena, B.R.; Kazemi, M.; et al. ASXL1 Mutations Are Associated with Distinct Epigenomic Alterations That Lead to Sensitivity to Venetoclax and Azacytidine. Blood Cancer J. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Shumilov, E.; Flach, J.; Kohlmann, A.; Banz, Y.; Bonadies, N.; Fiedler, M.; Pabst, T.; Bacher, U. Current Status and Trends in the Diagnostics of AML and MDS. Blood Rev. 2018, 32, 508–519. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Disease | Status | FLT3 | TP53 | Gene Variants | Karyotype |
|---|---|---|---|---|---|---|
| HL-60 | AML (M2) | de novo | wt | null | NRAS Q61L CDKN2A R80X | hypotetraploid |
| ML-2 | AML (M4) | de novo | wt | wt | KMT2A-AFDN KRAS A146T | t(6;11) |
| MOLM-13 | AML (M5) | relapse | ITD | wt | KMT2A-MLLT3 | t(9;11) |
| MOLM-16 | AML (M0) | relapse | wt | V173M/C238S | MLL V1368L | hypotetraploid |
| OCI-AML3 | AML (M4) | de novo | wt | wt | DNMT3A R882C NRAS Q61L NPM1 L287fs | +1, +5, +8 |
| PL-21 | AML (M3) | de novo | ITD/P336L | wt/P36fs | KRAS A146V | hypertetraploid |
| SKM-1 | AML (M5) | refractory | wt | R248Q/R248Q | ASXL1 Y591Ter KRAS K117N | del(9q12) |
| OCI-Ly1 | DLBCL | relapse | wt | R158H/C176G | BCL2-IgH, PTEN del | t(14;18) |
| Targeted Therapy | |||||||
|---|---|---|---|---|---|---|---|
| Cell Line | Venetoclax | A1331825 | PQR-309 | C-188-9 | PTC596 | S63845 | Trametinib |
| Target | BCL-2 | BCL-XL | PI3K, mTOR | STAT3 | BMI-1 | MCL-1 | MEK |
| HL-60 | 1 | 4 | 5 | 5 | 0.2 | 0.1 | 0.08 |
| ML-2 | 0.08 | 2 | 3 | 4 | 1.5 | 0.5 | 0.12 |
| MOLM-13 | 0.1 | 6 | 2 | 4 | 0.3 | 0.01 | 0.12 |
| MOLM-16 | >10 | 2 | 10 | >10 | 1.1 | 10 | 10 |
| OCI-AML3 | 0.2 | 4 | 10 | 8 | 0.5 | 0.2 | 0.1 |
| PL-21 | 10 | 2 | 10 | >10 | 0.8 | 1 | 10 |
| SKM-1 | 2 | 8 | 3 | 8 | 1.2 | 0.5 | 0.12 |
| OCI-Ly1 | 0.06 | 8 | 1 | 8 | 1 | 0.12 | 0.3 |
| Venetoclax Combination Treatment | ||||||
|---|---|---|---|---|---|---|
| Cell Line | A1331825 | Bimiralisib | C188-9 | PTC596 | S63845 | Trametinib |
| HL-60 | 0.6–0.8 | 0.2–0.4 | 0.4–0.6 | 0.2–0.4 | 0.4–0.6 | 0.2–0.4 |
| ML-2 | 0.3–0.5 | 0.3–0.5 | 0.9–1.1 | 0.8–1.0 | 0.1–0.3 | 0.3–0.5 |
| MOLM-13 | 0.2–0.4 | 0.3–0.5 | 0.9–1.1 | 0.6–0.8 | 0.2–0.4 | 0.2–0.4 |
| MOLM-16 | 0.2–0.4 | >1.1 | 0.3–0.5 | 0.7–0.9 | 0.7–0.9 | 0.9–1.1 |
| OCI-AML3 | 0.2–0.4 | 0.5–0.7 | 0.8–1.0 | 0.6–0.8 | 0.2–0.4 | 0.3–0.5 |
| PL-21 | 0.4–0.6 | 0.8–1.0 | 0.5–0.7 | 0.9–1.1 | >1.1 | 0.9–1.1 |
| SKM-1 | <0.1 | 0.3–0.5 | 0.7–0.9 | 0.3–0.5 | <0.1 | <0.1 |
| OCI-Ly1 | 0.4–0.6 | 0.6–0.8 | 0.7–0.9 | 0.8–1.0 | 0.4–0.6 | nd |
| ID | Disease | Mutation Profile | Cytogenetics | Source | PBC | BMI | CD34+ |
|---|---|---|---|---|---|---|---|
| % | % | % | |||||
| AML1 | AML-M1 | FLT3-ITD (>1), NPM1 | normal | PB | 90 | 90 | 5 |
| AML2 | AML-M1 | FLT3-ITD (0.78), U2AF1, BCOR, TET2 | del(20)(q11.2q13), +8 | PB | 62 | 90 | 18 |
| AML3 | AML-M5a | NPM1, IDH2 | normal | BM | 87 | 90 | 18 |
| AML4 | AML-M4 | FLT3-TKD, KMT2A-MLLT10 | t(10;11) | PB | 88 | 80 | 45 |
| AML5 | AML-M4 | NPM1, DNMT3A, NF1 | normal | BM | 8 | 20 | 11 |
| AML6 | AML-M5 | NPM1, FLT3-TKD (0.63), DNMT3A | normal | BM | 86 | 95 | 2 |
| AML7 | AML-M5 | NPM1, FLT3-ITD (0.58), DNMT3A | normal | PB | 1 | 70 | 7 |
| AML8 | AML sec | FLT3-ITD, IDH2, RUNX1, DNMT3A | tetraploid, del5q | BM | 45 | 80 | 56 |
| AML9 | AML-M1 | NPM1, FLT3-ITD (9.45), IDH2 | normal | PB | 94 | 90 | 20 |
| AML10 | AML-M4 | ASXL1, TET2, KRAS | normal | BM | nd | 30 | 1 |
| AML11 | AML-M4 | NPM1, PTPN11 | normal | BM | 65 | 65 | 22 |
| AML12 | AML-M5 | ASXL1, TET2, KRAS, SH2B3, U2AF1 | mono7 | PB | 53 | 80 | 30 |
| AML13 | AML sec | TET2, DNMT3A, PTPN11 | mono7, del(12), inv(9) | PB | 36 | 50 | 90 |
| AML14 | MDS-AML | CEBPA, ASXL1, EZH2, RUNX1 | normal | BM | 13 | nd | 26 |
| AML15 | MDS-AML | ASXL1, TP53, CALR | KMT2A amp (97%) | PB | 20 | 20 | 52 |
| AML16 | AML-M1 | normal | mono9, 11q23.3 | PB | 96 | 90 | 38 |
| AML17 | AML-M4/5 | NPM1, DNMT3A, TET2, PTPN11 | normal | PB | 19 | 90 | 68 |
| AML18 | AML-M1 | NPM1, IDH2, SRSF2 | normal | PB | 72 | 70 | 1 |
| AML19 | AML-M2 | IDH2, DNMT3A | der(16)t(11;16), +14 | BM | 33 | 60 | 90 |
| AML20 | AML-M4 | ASXL1, IDH2, DNMT3A, SRSF2 | normal, +8 | BM | 9 | 50 | 82 |
| AML21 | AML-M2 | NPM1, IDH2 | normal | BM | 95 | 90 | 24 |
| AML22 | AML-M0 | ASXL1, IDH2, RUNX1 | normal | BM | 64 | 80 | 94 |
| AML23 | AML-M2 | RUNX1, TET2, PTPN11, PRPF8, NF1 | mono7, t(9;22) | PB | 68 | 45 | 82 |
| AML24 | AML sec | FLT3-TKD, IDH1, NPM1, PTPN11, SRSF2 | normal | BM | 83 | 90 | 1 |
| AML25 | AML-M1 | TP53 | complex | PB | 75 | 80 | 97 |
| AML26 | AML-M4 | FLT3-TKD (0.56), TET2, SRFS2, TP53 | normal, +8 | PB | 46 | 50 | 10 |
| CML1 | CML | BCR-ABL1 | t(9;22) | PB | 1 | 5 | 42 |
| NHL1 | NHL | TP53 | normal | PB | nd | nd | nd |
| NHL2 | NHL | cMyc and BCL2 rearranged (double hit) | t(8;14), t(14;18) | BM | 18 | 89 | 58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seipel, K.; Brügger, Y.; Mandhair, H.; Bacher, U.; Pabst, T. Rationale for Combining the BCL2 Inhibitor Venetoclax with the PI3K Inhibitor Bimiralisib in the Treatment of IDH2- and FLT3-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 12587. https://doi.org/10.3390/ijms232012587
Seipel K, Brügger Y, Mandhair H, Bacher U, Pabst T. Rationale for Combining the BCL2 Inhibitor Venetoclax with the PI3K Inhibitor Bimiralisib in the Treatment of IDH2- and FLT3-Mutated Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2022; 23(20):12587. https://doi.org/10.3390/ijms232012587
Chicago/Turabian StyleSeipel, Katja, Yvo Brügger, Harpreet Mandhair, Ulrike Bacher, and Thomas Pabst. 2022. "Rationale for Combining the BCL2 Inhibitor Venetoclax with the PI3K Inhibitor Bimiralisib in the Treatment of IDH2- and FLT3-Mutated Acute Myeloid Leukemia" International Journal of Molecular Sciences 23, no. 20: 12587. https://doi.org/10.3390/ijms232012587
APA StyleSeipel, K., Brügger, Y., Mandhair, H., Bacher, U., & Pabst, T. (2022). Rationale for Combining the BCL2 Inhibitor Venetoclax with the PI3K Inhibitor Bimiralisib in the Treatment of IDH2- and FLT3-Mutated Acute Myeloid Leukemia. International Journal of Molecular Sciences, 23(20), 12587. https://doi.org/10.3390/ijms232012587