
TLR4-Pathway-Associated Biomarkers in Subarachnoid Hemorrhage (SAH): Potential Targets for Future Anti-Inflammatory Therapies
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Microglia as Key Players of the CNS Innate Immune System
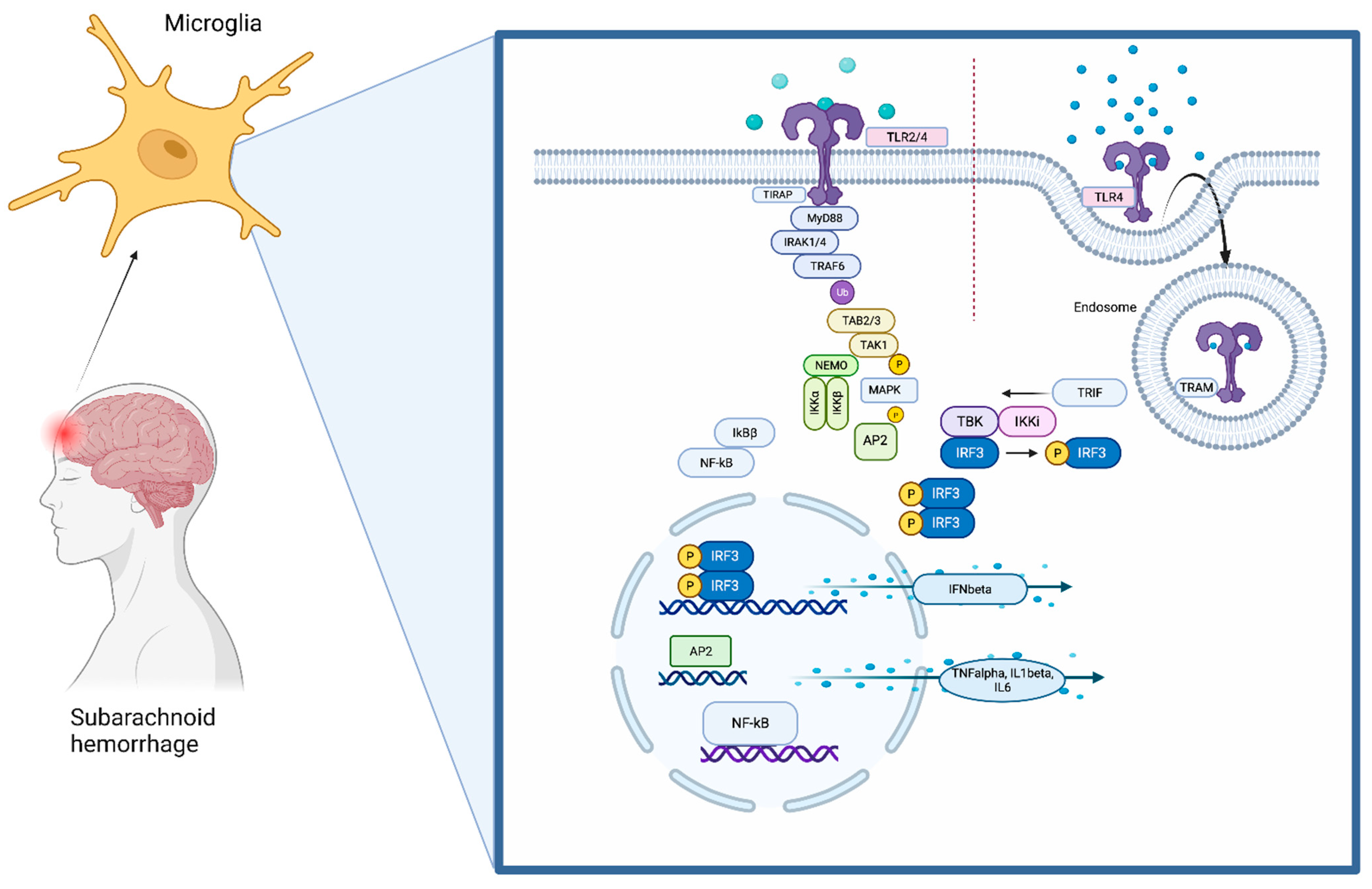
3. The TLR4 Pathway
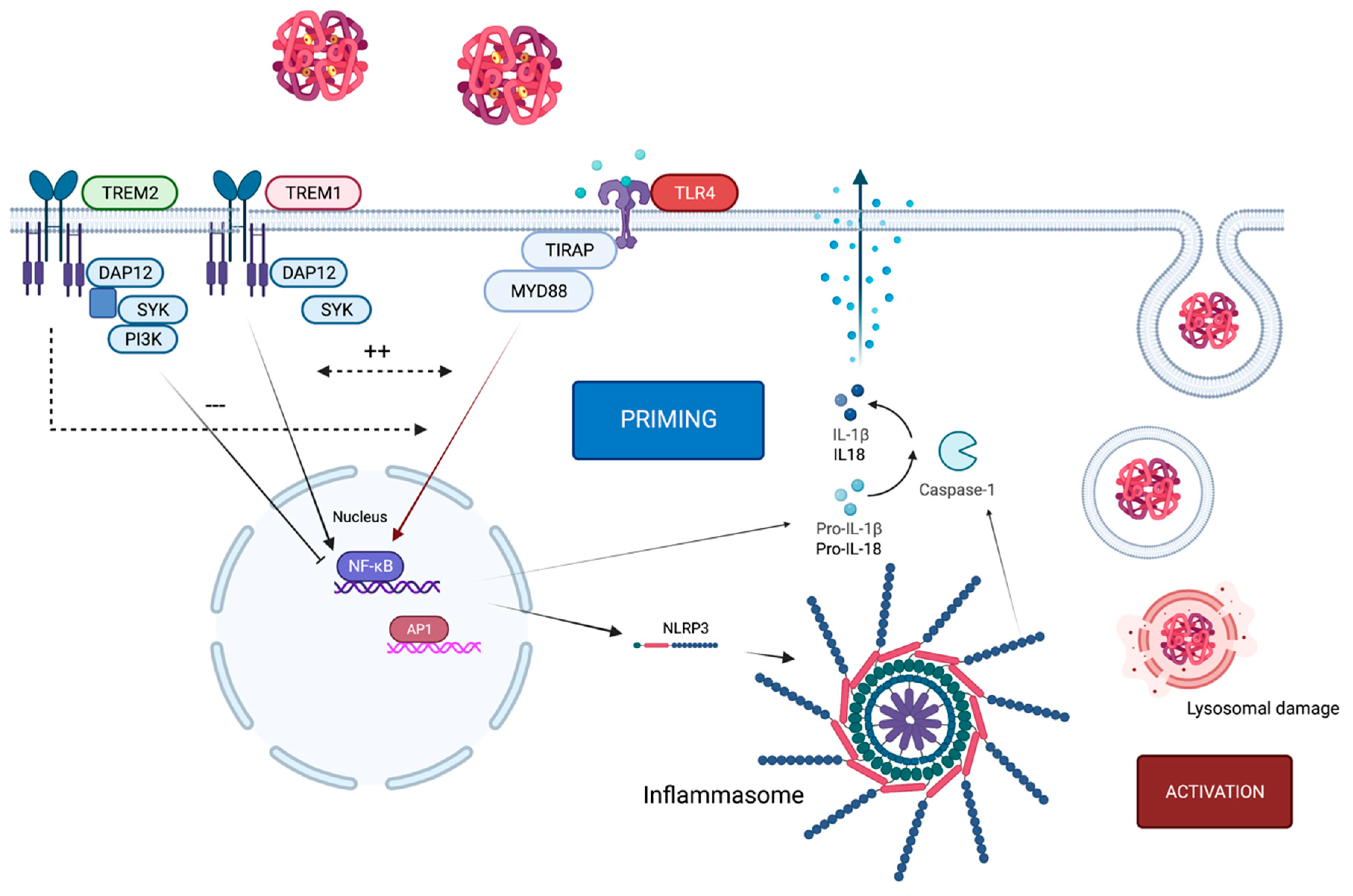
4. Crosstalk of TLR4 and the NLRP3 Inflammasome
5. Co-Existence of TLR4 Alongside TREM1/2
6. Pathophysiology of Inflammation after SAH
7. TLR4-Pathway-Associated Proteins Acting as Biomarkers following SAH
7.1. Ligands of TLR4
7.1.1. HMGB1
7.1.2. Red Blood Cell Lysate
7.2. Inflammatory Cytokines
7.3. TLR4
7.4. TREM1
7.5. Inflammasome Proteins
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SAH | subarachnoid hemorrhage |
| TLR4 | Toll-like receptor 4 |
| CNS | central nervous system |
| BBB | blood–brain barrier |
| DAMP | danger-associated molecular pattern |
| PAMP | pathogen-associated molecular pattern |
| PRR | pattern recognition receptor |
| MyD88 | myeloid differentiation primary response protein 88 (MyD88) |
| TRIF | TIR-domain-containing adaptor-inducing IFN-beta |
| MAPK | mitogen-activated protein kinase |
| TREM | triggering receptor expressed on myeloid cells |
| HMGB1 | high mobility group box 1 |
| NLRP3 | NLR family pyrin domain containing 3 |
| ICAM1 | intercellular adhesion molecule 1 |
| ROS | reactive oxygen species |
| COX 2 | Cyclooxygenase 2 |
| TREM | triggering receptor expressed on myeloid cells |
References
- Schneider, U.C.; Davids, A.M.; Brandenburg, S.; Muller, A.; Elke, A.; Magrini, S.; Atangana, E.; Turkowski, K.; Finger, T.; Gutenberg, A.; et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015, 130, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A., Jr. Decoding the patterns of self and nonself by the innate immune system. Science 2002, 296, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsan, M.F.; Gao, B. Endogenous ligands of Toll-like receptors. J. Leukoc. Biol. 2004, 76, 514–519. [Google Scholar] [CrossRef]
- Pineau, I.; Lacroix, S. Endogenous signals initiating inflammation in the injured nervous system. Glia 2009, 57, 351–361. [Google Scholar] [CrossRef]
- Balanca, B.; Desmurs, L.; Grelier, J.; Perret-Liaudet, A.; Lukaszewicz, A.C. DAMPs and RAGE Pathophysiology at the Acute Phase of Brain Injury: An Overview. Int. J. Mol. Sci. 2021, 22, 2439. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Tang, J.; Yang, X.; Huang, J.; Zhu, T.; Zhao, F.; Li, S.; Li, X.; Qu, Y.; et al. Role of toll-like receptor 4 in the regulation of the cell death pathway and neuroinflammation. Brain Res. Bull 2019, 148, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 2021, 91, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhou, W.; Yan, Z.; Qu, M.; Bu, X. Toll-like receptor 4 (TLR4) is correlated with delayed cerebral ischemia (DCI) and poor prognosis in aneurysmal subarachnoid hemorrhage. J. Neurol. Sci. 2015, 359, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Suzuki, H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regen. Res. 2017, 12, 193–196. [Google Scholar] [PubMed]
- Weiland, J.; Beez, A.; Westermaier, T.; Kunze, E.; Siren, A.L.; Lilla, N. Neuroprotective Strategies in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2021, 22, 5542. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Q.; Zhang, Q.; Lu, Y.; Liu, J.; Li, W.; Lv, S.; Zhou, M.; Zhang, X.; Hang, C. Resveratrol Attenuates Early Brain Injury after Experimental Subarachnoid Hemorrhage via Inhibition of NLRP3 Inflammasome Activation. Front. Neurosci. 2017, 11, 611. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, S.R.; Hafez, A.; Jahromi, B.R.; Kinfe, T.M.; Lamprecht, A.; Niemela, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; EI Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Almad, A.; Maragakis, N.J. A stocked toolbox for understanding the role of astrocytes in disease. Nat. Rev. Neurol. 2018, 14, 351–362. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, R.; Brandenburg, S.; Nieminen-Kelha, M.; Kremenetskaia, I.; Boehm-Sturm, P.; Vajkoczy, P.; Schneider, C.U. Microglia as target for anti-inflammatory approaches to prevent secondary brain injury after subarachnoid hemorrhage (SAH). J. Neuroinflammation 2021, 18, 36. [Google Scholar] [CrossRef]
- Khey, K.M.W.; Huard, A.; Mahmoud, S.H. Inflammatory Pathways Following Subarachnoid Hemorrhage. Cell Mol. Neurobiol. 2020, 40, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflammation 2013, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Palsson-McDermott, E.M.; Doyle, S.L.; McGettrick, A.F.; Hardy, M.; Husebye, H.; Banahan, K.; Gong, M.; Golenbock, D.; Espevik, T.; O’Neill, L.A. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat. Immunol. 2009, 10, 579–586. [Google Scholar] [CrossRef]
- Tseng, P.H.; Matsuzawa, A.; Zhang, W.; Mino, T.; Vignali, D.A.; Karin, M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 2010, 11, 70–75. [Google Scholar] [CrossRef]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Huai, W.; Song, H.; Wang, L.; Li, B.; Zhao, J.; Han, L.; Gao, C.; Jiang, G.; Zhang, L.; Zhao, W. Phosphatase PTPN4 preferentially inhibits TRIF-dependent TLR4 pathway by dephosphorylating TRAM. J. Immunol. 2015, 194, 4458–4465. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hong, Y.; Xie, Y.; Yuan, K.; Li, J.; Sun, R.; Zhang, X.; Shi, X.; Li, R.; Wu, J.; et al. TREM-1 Exacerbates Neuroinflammatory Injury via NLRP3 Inflammasome-Mediated Pyroptosis in Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2020, 12, 643–659. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Tsuruta, R.; Kaneko, T.; Yamashita, S.; Fujita, M.; Kasaoka, S.; Hashiguchi, T.; Suzuki, M.; Maruyama, I.; Maekawa, T. High-mobility group box 1 protein in CSF of patients with subarachnoid hemorrhage. Neurocrit. Care 2009, 11, 362–368. [Google Scholar] [CrossRef]
- Zhong, W.J.; Duan, J.X.; Liu, T.; Yang, H.H.; Guan, X.X.; Zhang, C.Y.; Yang, J.T.; Xiong, J.B.; Zhou, Y.; Guan, C.X.; et al. Activation of NLRP3 inflammasome up-regulates TREM-1 expression in murine macrophages via HMGB1 and IL-18. Int. Immunopharmacol. 2020, 89 Pt A, 107045. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Forster, I.; Farlik, M.; Decker, T.; Pasquier, R.A.D.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Shao, G.F.; Chen, J.L.; Gong, J. The NLRP3 Inflammasome: An Important Driver of Neuroinflammation in Hemorrhagic Stroke. Cell Mol. Neurobiol. 2018, 38, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Dong, Q.; Song, Z.; Shen, F.; Shi, J.; Li, Y. NLRP3 inflammasome: A promising target in ischemic stroke. Inflamm. Res. 2017, 66, 17–24. [Google Scholar] [CrossRef]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. TREM-1: A new regulator of innate immunity in sepsis syndrome. Nat. Med. 2001, 7, 530–532. [Google Scholar] [CrossRef]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Zhang, Y.D.; Gao, Q.; Zhou, J.S.; Zhu, X.C.; Lu, H.; Shi, J.Q.; Tan, L.; Chen, Q.; Yu, J.T. TREM1 facilitates microglial phagocytosis of amyloid beta. Acta Neuropathol. 2016, 132, 667–683. [Google Scholar] [CrossRef]
- Replogle, J.M.; Chan, G.; White, C.C.; Raj, T.; Winn, P.A.; Evans, D.A.; Sperling, R.A.; Chibnik, L.B.; Bradshaw, E.M.; Schneider, J.A.; et al. A TREM1 variant alters the accumulation of Alzheimer-related amyloid pathology. Ann. Neurol. 2015, 77, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Zhang, X.; Liu, Q.; Xie, Y.; Shi, X.; Chen, J.; Li, Y.; Guo, H.; Sun, R.; Hong, Y.; et al. Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. 2019, 10, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Peng, J.; Sherchan, P.; Ma, Y.; Xiang, S.; Yan, F.; Zhao, H.; Jiang, Y.; Wang, N.; Zhang, J.H.; et al. TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J. Neuroinflammation 2020, 17, 168. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef]
- Hamerman, J.A.; Tchao, N.K.; Lowell, C.A.; Lanier, L.L. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat. Immunol. 2005, 6, 579–586. [Google Scholar] [CrossRef]
- Long, H.; Zhong, G.; Wang, C.; Zhang, J.; Zhang, Y.; Luo, J.; Shi, S. TREM2 Attenuates Abeta1-42-Mediated Neuroinflammation in BV-2 Cells by Downregulating TLR Signaling. Neurochem. Res. 2019, 44, 1830–1839. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Schweizer, T.A. Spontaneous subarachnoid haemorrhage. Lancet 2017, 389, 655–666. [Google Scholar] [CrossRef]
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, R.L.; Higashida, R.T.; Keller, E.; Mayer, S.A.; Molyneux, A.; Raabe, A.; Vajkoczy, P.; Wanke, I.; Bach, D.; Frey, A.; et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: A randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011, 10, 618–625. [Google Scholar] [CrossRef]
- Cahill, J.; Zhang, J.H. Subarachnoid hemorrhage: Is it time for a new direction? Stroke 2009, 40 (Suppl. 3), S86–S87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H. Inflammation: A Good Research Target to Improve Outcomes of Poor-Grade Subarachnoid Hemorrhage. Transl. Stroke Res. 2019, 10, 597–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira Manoel, A.L.; Macdonald, R.L. Neuroinflammation as a Target for Intervention in Subarachnoid Hemorrhage. Front. Neurol. 2018, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Lucke-Wold, B.P.; Logsdon, A.F.; Manoranjan, B.; Turner, R.C.; McConnell, E.; Vates, G.E.; Huber, J.D.; Rosen, C.L.; Simard, J.M. Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review. Int. J. Mol. Sci. 2016, 17, 497. [Google Scholar] [CrossRef] [Green Version]
- Gris, T.; Laplante, P.; Thebault, P.; Cayrol, R.; Najjar, A.; Joannette-Pilon, B.; Brillant-Marquis, F.; Magro, E.; English, S.W.; Lapointe, R.; et al. Innate immunity activation in the early brain injury period following subarachnoid hemorrhage. J. Neuroinflammation 2019, 16, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atangana, E.; Schneider, U.C.; Blecharz, K.; Magrini, S.; Wagner, J.; Nieminen-Kelha, M.; Kremenetskaia, I.; Heppner, F.L.; Engelhardt, B.; Vajkoczy, P. Intravascular Inflammation Triggers Intracerebral Activated Microglia and Contributes to Secondary Brain Injury After Experimental Subarachnoid Hemorrhage (eSAH). Transl. Stroke Res. 2017, 8, 144–156. [Google Scholar] [CrossRef]
- Fang, Y.; Gao, S.; Wang, X.; Cao, Y.; Lu, J.; Chen, S.; Lenahan, C.; Zhang, J.H.; Shao, A.; Zhang, J. Programmed Cell Deaths and Potential Crosstalk With Blood-Brain Barrier Dysfunction After Hemorrhagic Stroke. Front. Cell Neurosci. 2020, 14, 68. [Google Scholar] [CrossRef] [Green Version]
- Blecharz-Lang, K.G.; Wagner, J.; Fries, A.; Nieminen-Kelha, M.; Rosner, J.; Schneider, U.C.; Vajkoczy, P. Interleukin 6-Mediated Endothelial Barrier Disturbances Can Be Attenuated by Blockade of the IL6 Receptor Expressed in Brain Microvascular Endothelial Cells. Transl. Stroke Res. 2018, 9, 631–642. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Pagan, V.A.; Hanafy, K.A. The role of TLR4 and HO-1 in neuroinflammation after subarachnoid hemorrhage. J. Neurosci. Res. 2020, 98, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Gallia, G.L.; Tamargo, R.J. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 750–758. [Google Scholar] [CrossRef]
- Chen, J.; Wang, L.; Xu, H.; Xing, L.; Zhuang, Z.; Zheng, Y.; Li, X.; Wang, C.; Chen, S.; Guo, Z.; et al. Meningeal lymphatics clear erythrocytes that arise from subarachnoid hemorrhage. Nat. Commun. 2020, 11, 3159. [Google Scholar] [CrossRef] [PubMed]
- Pradilla, G.; Chaichana, K.L.; Hoang, S.; Huang, J.; Tamargo, R.J. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg. Clin. N. Am. 2010, 21, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.C.; Schiffler, J.; Hakiy, N.; Horn, P.; Vajkoczy, P. Functional analysis of Pro-inflammatory properties within the cerebrospinal fluid after subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflammation 2012, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.H.; Savarraj, J.P.J.; Parsha, K.; Hergenroeder, G.W.; Chang, T.R.; Kim, D.H.; Kitagawa, R.S.; Blackburn, S.L.; Choi, H.A. Inflammation in delayed ischemia and functional outcomes after subarachnoid hemorrhage. J. Neuroinflammation 2019, 16, 213. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, K.; Ikedo, T.; Kamio, Y.; Furukawa, H.; Lawton, M.T.; Hashimoto, T. TLR4 (Toll-Like Receptor 4) Mediates the Development of Intracranial Aneurysm Rupture. Hypertension 2020, 75, 468–476. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Q.; Xiong, X.Y.; Gong, Q.W.; Liao, M.F.; Yang, Q.W. TLR4 gene polymorphisms rs11536889 is associated with intra cranial aneurysm susceptibility. J. Clin. Neurosci. 2018, 53, 165–170. [Google Scholar] [CrossRef]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Hemmer, S.; Senger, S.; Griessenauer, C.J.; Simgen, A.; Oertel, J.; Geisel, J.; Hendrix, P. Admission serum high mobility group box 1 (HMGB1) protein predicts delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 2022, 45, 807–817. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Guresir, A.; Stoffel-Wagner, B.; Fimmers, R.; Kinfe, T.M.; Dietrich, D.; Lamprecht, A.; Vatter, H.; Guresir, E.; Muhammad, S. Systemic High-Mobility Group Box-1: A Novel Predictive Biomarker for Cerebral Vasospasm in Aneurysmal Subarachnoid Hemorrhage. Crit. Care Med. 2018, 46, e1023–e1028. [Google Scholar] [CrossRef]
- Zhao, X.D.; Mao, H.Y.; Lv, J.; Lu, X.J. Expression of high-mobility group box-1 (HMGB1) in the basilar artery after experimental subarachnoid hemorrhage. J. Clin. Neurosci. 2016, 27, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Chen, J.S.; Zhou, F.; Liu, Q.C.; Chen, G.; Zhang, J.M. Relationship between plasma high mobility group box-1 protein levels and clinical outcomes of aneurysmal subarachnoid hemorrhage. J. Neuroinflammation 2012, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ieong, C.; Sun, H.; Wang, Q.; Ma, J. Glycyrrhizin suppresses the expressions of HMGB1 and ameliorates inflammative effect after acute subarachnoid hemorrhage in rat model. J. Clin. Neurosci. 2018, 47, 278–284. [Google Scholar] [CrossRef]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef] [Green Version]
- Gram, M.; Sveinsdottir, S.; Ruscher, K.; Hansson, S.R.; Cinthio, M.; Akerstrom, B.; Ley, D. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J. Neuroinflammation 2013, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Tang, S.C.; Lee, J.E.; Lai, D.M.; Huang, S.J.; Hsieh, S.T.; Jeng, J.S.; Tu, Y.K. Prognostic value of intrathecal heme oxygenase-1 concentration in patients with Fisher Grade III aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2014, 121, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Frase, S.; Steimer, M.; Selzner, L.; Kaiser, S.; Foit, N.A.; Niesen, W.D.; Schallner, N. Temporal Expression Pattern of Hemoxygenase-1 Expression and Its Association with Vasospasm and Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. Neurocrit. Care 2022, 36, 279–291. [Google Scholar] [CrossRef]
- Bozza, M.T.; Jeney, V. Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front. Immunol. 2020, 11, 1323. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Ridwan, S.; Grote, A.; Simon, M. Interleukin 6 in cerebrospinal fluid is a biomarker for delayed cerebral ischemia (DCI) related infarctions after aneurysmal subarachnoid hemorrhage. Sci. Rep. 2021, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Lenski, M.; Huge, V.; Briegel, J.; Tonn, J.C.; Schichor, C.; Thon, N. Interleukin 6 in the Cerebrospinal Fluid as a Biomarker for Onset of Vasospasm and Ventriculitis After Severe Subarachnoid Hemorrhage. World Neurosurg. 2017, 99, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Guan, Y.; Zhao, G.; Fu, X.J.; Guo, T.Z.; Liu, Y.T.; Ren, X.L.; Wang, W.; Liu, H.R.; Li, Y.Q. Elevated IL-6 and TNF-alpha Levels in Cerebrospinal Fluid of Subarachnoid Hemorrhage Patients. Mol. Neurobiol. 2016, 53, 3277–3285. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Stuart, R.M.; Khandji, A.G.; Connolly, E.S.; Badjatia, N.; Mayer, S.A.; Schindler, C. Relationship between brain interstitial fluid tumor necrosis factor-alpha and cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J. Clin. Neurosci. 2010, 17, 853–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, J.; Ogungbenro, K.; Hulme, S.; Patel, H.; Scarth, S.; Hoadley, M.; Illingworth, K.; McMahon, C.J.; Tzerakis, N.; King, A.T.; et al. Reduction of inflammation after administration of interleukin-1 receptor antagonist following aneurysmal subarachnoid hemorrhage: Results of the Subcutaneous Interleukin-1Ra in SAH (SCIL-SAH) study. J. Neurosurg. 2018, 128, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Hopkins, S.J.; Hulme, S.; Galea, J.P.; Hoadley, M.; Vail, A.; Hutchinson, P.J.; Grainger, S.; Rothwell, N.J.; King, A.T.; et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: A phase II randomised controlled trial. J. Neuroinflammation 2014, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Sarrafzadeh, A.; Schlenk, F.; Gericke, C.; Vajkoczy, P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2010, 13, 339–346. [Google Scholar] [CrossRef]
- Smith, C.J.; Hulme, S.; Vail, A.; Heal, C.; Parry-Jones, A.R.; Scarth, S.; Hopkins, K.; Hoadley, M.; Allan, S.M.; Rothwell, N.J.; et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke 2018, 49, 1210–1216. [Google Scholar] [CrossRef] [Green Version]
- Sokol, B.; Wasik, N.; Jankowski, R.; Holysz, M.; Wieckowska, B.; Jagodzinski, P. Soluble Toll-Like Receptors 2 and 4 in Cerebrospinal Fluid of Patients with Acute Hydrocephalus following Aneurysmal Subarachnoid Haemorrhage. PLoS ONE 2016, 11, e0156171. [Google Scholar]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Kawakita, F.; Nishikawa, H.; Nakano, F.; Liu, L.; Suzuki, H. Selective Toll-Like Receptor 4 Antagonists Prevent Acute Blood-Brain Barrier Disruption After Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2019, 56, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Lei, L.; Nishikawa, H.; Nakano, F.; Nakatsuka, Y.; Suzuki, H. TAK-242, Toll-Like Receptor 4 Antagonist, Attenuates Brain Edema in Subarachnoid Hemorrhage Mice. Acta Neurochir. Suppl. 2020, 127, 77–81. [Google Scholar] [PubMed]
- Liu, F.Y.; Cai, J.; Wang, C.; Ruan, W.; Guan, G.P.; Pan, H.Z.; Li, J.R.; Qian, C.; Chen, J.S.; Wang, L.; et al. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: A possible role for the regulation of TLR4/MyD88/NF-kappaB signaling pathway. J. Neuroinflammation 2018, 15, 347. [Google Scholar] [CrossRef] [PubMed]
- Rahimifard, M.; Maqbool, F.; Moeini-Nodeh, S.; Niaz, K.; Abdollahi, M.; Braidy, N.; Nabavi, S.M.; Nabavi, S.F. Targeting the TLR4 signaling pathway by polyphenols: A novel therapeutic strategy for neuroinflammation. Ageing Res. Rev. 2017, 36, 11–19. [Google Scholar] [CrossRef]
- Lee, J.W.; Ahn, J.Y.; Hasegawa, S.; Cha, B.Y.; Yonezawa, T.; Nagai, K.; Seo, H.J.; Jeon, W.B.; Woo, J.T. Inhibitory effect of luteolin on osteoclast differentiation and function. Cytotechnology 2009, 61, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Tan, X.; Xu, J.; Wang, T.; Liang, T.; Xu, X.; Ma, C.; Xu, Z.; Wang, W.; Li, H.; et al. Luteolin alleviates neuroinflammation via downregulating the TLR4/TRAF6/NF-kappaB pathway after intracerebral hemorrhage. Biomed Pharmacother. 2020, 126, 110044. [Google Scholar] [CrossRef]
- Park, S.J.; Song, H.Y.; Youn, H.S. Suppression of the TRIF-dependent signaling pathway of toll-like receptors by isoliquiritigenin in RAW264.7 macrophages. Mol. Cells 2009, 28, 365–368. [Google Scholar] [CrossRef]
- Zhu, H.T.; Bian, C.; Yuan, J.C.; Chu, W.H.; Xiang, X.; Chen, F.; Wang, C.S.; Feng, H.; Lin, J.K. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J. Neuroinflammation 2014, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.G.; Ma, Q.; Jing, G.; Wang, G.Q.; Hao, X.D.; Wang, L. Increased levels of soluble triggering receptor expressed on myeloid cells-1 in cerebrospinal fluid of subarachnoid hemorrhage patients. J. Clin. Neurosci. 2017, 35, 139–143. [Google Scholar] [CrossRef]
- Hirsch, Y.; Geraghty, J.R.; Katz, E.A.; Testai, F.D. Inflammasome Caspase-1 Activity is Elevated in Cerebrospinal Fluid After Aneurysmal Subarachnoid Hemorrhage and Predicts Functional Outcome. Neurocrit. Care 2021, 34, 889–898. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, X.L.; Yu, Q.; Pan, H.; Zhang, X.S.; Zhang, Q.R.; Wang, H.D.; Zhang, X. Inflammasome Proteins in Cerebrospinal Fluid of Patients with Subarachnoid Hemorrhage are Biomarkers of Early Brain Injury and Functional Outcome. World Neurosurg. 2016, 94, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018, 8, 5971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Chen, H.; Jin, J.; Liu, Q.; Zhong, D.; Li, G. Inhibition of the NLRP3 inflammasome reduces brain edema and regulates athe distribution of aquaporin-4 after cerebral ischaemia-reperfusion. Life Sci. 2020, 251, 117638. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinz, R.; Schneider, U.C. TLR4-Pathway-Associated Biomarkers in Subarachnoid Hemorrhage (SAH): Potential Targets for Future Anti-Inflammatory Therapies. Int. J. Mol. Sci. 2022, 23, 12618. https://doi.org/10.3390/ijms232012618
Heinz R, Schneider UC. TLR4-Pathway-Associated Biomarkers in Subarachnoid Hemorrhage (SAH): Potential Targets for Future Anti-Inflammatory Therapies. International Journal of Molecular Sciences. 2022; 23(20):12618. https://doi.org/10.3390/ijms232012618
Chicago/Turabian StyleHeinz, Rebecca, and Ulf C. Schneider. 2022. "TLR4-Pathway-Associated Biomarkers in Subarachnoid Hemorrhage (SAH): Potential Targets for Future Anti-Inflammatory Therapies" International Journal of Molecular Sciences 23, no. 20: 12618. https://doi.org/10.3390/ijms232012618
APA StyleHeinz, R., & Schneider, U. C. (2022). TLR4-Pathway-Associated Biomarkers in Subarachnoid Hemorrhage (SAH): Potential Targets for Future Anti-Inflammatory Therapies. International Journal of Molecular Sciences, 23(20), 12618. https://doi.org/10.3390/ijms232012618