

Lack of NPR1 Increases Vascular Endothelial Adhesion through Induction of Integrin Beta 4

 , ,
, ,  and
and 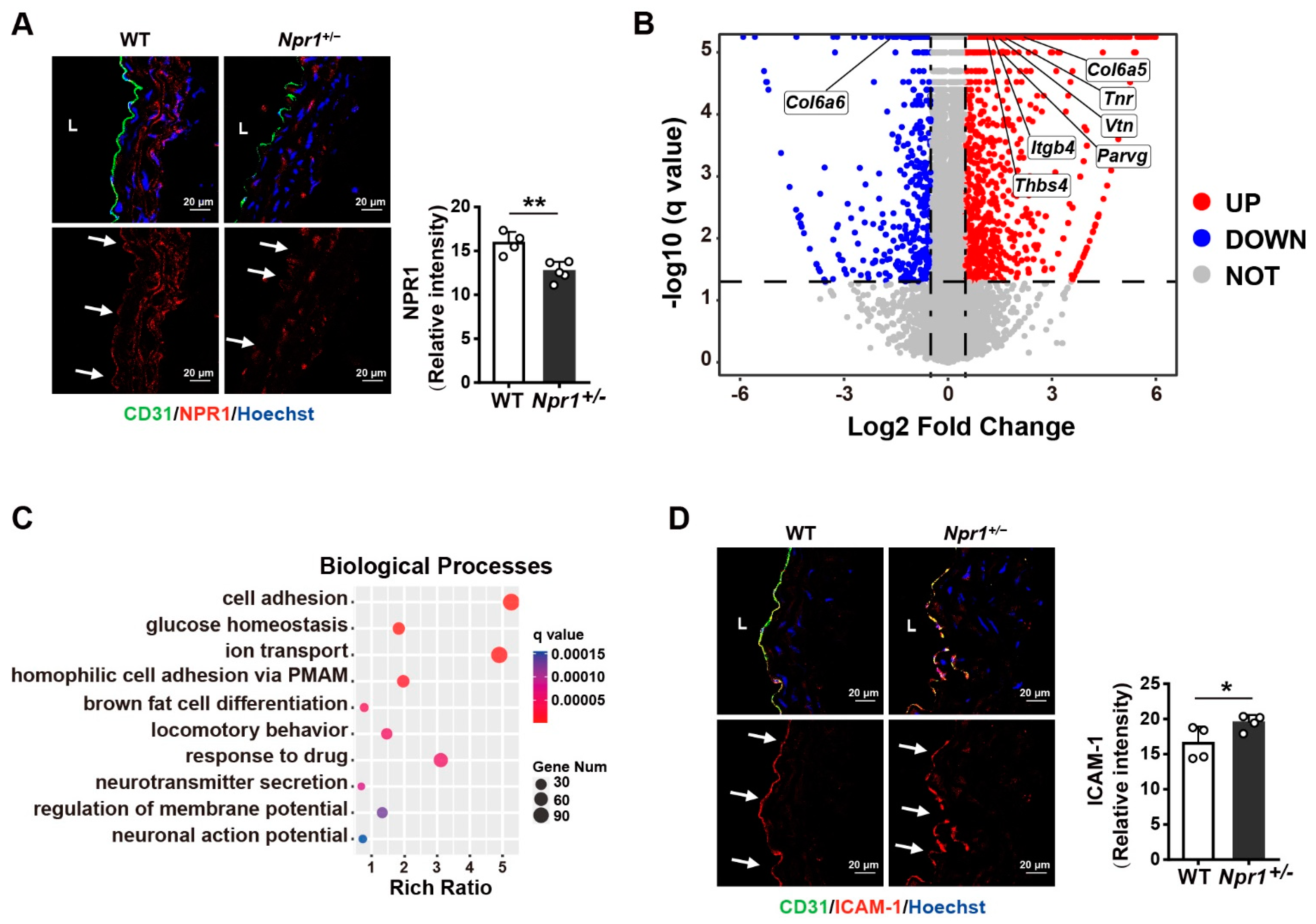{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. NPR1 Deficiency Increases Vascular Endothelial Cell Adhesion
2.2. Endothelial Cell Adhesion Is Enhanced by Knockdown of NPR1 but Reduced by Overexpression of NPR1
2.3. Integrin Beta 4 (Itgb4) Is Identified as a Key Gene Related to Focal Adhesion under NPR1 Deficiency
2.4. NPR1 Suppresses Monocyte–Endothelial Cell Adhesion by Reducing ITGB4 Expression
2.5. Atherosclerosis Mouse Model Exhibits Decreased NPR1 and Increased ITGB4 Expression
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Mouse Models
4.3. Total RNA Extraction
4.4. Real-Time Quantitative PCR (qPCR)
4.5. RNA-Sequencing Analysis
4.6. siRNA Transfection
4.7. Overexpression of NPR1 in Tumor Necrosis Factor-α (TNF-α)-Treated HUVECs
4.8. Immunofluorescent Staining
4.9. Western Blot
4.10. Monocyte-Endothelial Cell Adhesion Assay
4.11. Oil Red O Staining
4.12. Measurement of cGMP Production
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NPR1 | natriuretic peptide receptor 1 |
| ICAM-1 | intercellular adhesion molecule 1 |
| ITGB4 | integrin beta 4 |
| ANP | atrial natriuretic peptide |
| cGMP | cyclic guanosine monophosphate |
| TNF-α | tumor necrosis factor-α |
| Tnr | tenascin R |
| Parvg | parvin gamma |
| Col6a6 | collagen type VI alpha 6 chain |
| Col6a5 | collagen type VI alpha 5 chain |
| Thbs4 | thrombospondin 4 |
| Vtn | vitronectin |
References
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [Green Version]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Čejková, S.; Králová Lesná, I.; Poledne, R. Monocyte adhesion to the endothelium is an initial stage of atherosclerosis development. Cor Vasa 2016, 58, e419–e425. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Lusis, A.J.; Fogelman, A.M.; Fonarow, G.C. Genetic basis of atherosclerosis: Part I: New genes and pathways. Circulation 2004, 110, 1868–1873. [Google Scholar] [CrossRef]
- Wang, L.; Dong, Z.; Zhang, Y.; Miao, J. The roles of integrin beta4 in vascular endothelial cells. J. Cell Physiol. 2012, 227, 474–478. [Google Scholar] [CrossRef]
- Tibaut, M.; Caprnda, M.; Kubatka, P.; Sinkovic, A.; Valentova, V.; Filipova, S.; Gazdikova, K.; Gaspar, L.; Mozos, I.; Egom, E.E.; et al. Markers of Atherosclerosis: Part 1—Serological Markers. Heart Lung Circ. 2019, 28, 667–677. [Google Scholar] [CrossRef]
- Alexander, M.R.; Knowles, J.W.; Nishikimi, T.; Maeda, N. Increased atherosclerosis and smooth muscle cell hypertrophy in natriuretic peptide receptor A-/-apolipoprotein E-/- mice. Arter. Thromb. Vasc. Biol. 2003, 23, 1077–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennel, S.J.; Godfrey, V.; Ch’ang, L.Y.; Lankford, T.K.; Foote, L.J.; Makkinje, A. The beta 4 subunit of the integrin family is displayed on a restricted subset of endothelium in mice. J. Cell Sci. 1992, 101 Pt 1, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Welser-Alves, J.V.; Boroujerdi, A.; Tigges, U.; Wrabetz, L.; Feltri, M.L.; Milner, R. Endothelial beta4 integrin is predominantly expressed in arterioles, where it promotes vascular remodeling in the hypoxic brain. Arter. Thromb. Vasc. Biol. 2013, 33, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Liu, X.; Qi, L.; Xu, J.; Zhao, J.; Zhang, Y.; Zhang, S.; Miao, J. Modulation of vascular endothelial cell senescence by integrin beta4. J. Cell Physiol. 2010, 225, 673–681. [Google Scholar] [CrossRef]
- Habas, K.; Shang, L. Alterations in intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in human endothelial cells. Tissue Cell 2018, 54, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [Green Version]
- Caprio, M.; Newfell, B.G.; la Sala, A.; Baur, W.; Fabbri, A.; Rosano, G.; Mendelsohn, M.E.; Jaffe, I.Z. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ. Res. 2008, 102, 1359–1367. [Google Scholar] [CrossRef] [Green Version]
- da Silva, J.F.; Alves, J.V.; Silva-Neto, J.A.; Costa, R.M.; Neves, K.B.; Alves-Lopes, R.; Carmargo, L.L.; Rios, F.J.; Montezano, A.C.; Touyz, R.M.; et al. Lysophosphatidylcholine induces oxidative stress in human endothelial cells via NOX5 activation—Implications in atherosclerosis. Clin. Sci. 2021, 135, 1845–1858. [Google Scholar] [CrossRef]
- Ding, Y.; Huang, L.; Xian, X.; Yuhanna, I.S.; Wasser, C.R.; Frotscher, M.; Mineo, C.; Shaul, P.W.; Herz, J. Loss of Reelin protects against atherosclerosis by reducing leukocyte-endothelial cell adhesion and lesion macrophage accumulation. Sci. Signal. 2016, 9, ra29. [Google Scholar] [CrossRef] [Green Version]
- Nishikimi, T.; Inaba-Iemura, C.; Ishimura, K.; Tadokoro, K.; Koshikawa, S.; Ishikawa, K.; Akimoto, K.; Hattori, Y.; Kasai, K.; Minamino, N.; et al. Natriuretic peptide/natriuretic peptide receptor-A (NPR-A) system has inhibitory effects in renal fibrosis in mice. Regul. Pept. 2009, 154, 44–53. [Google Scholar] [CrossRef]
- Pandey, K.N. Molecular and genetic aspects of guanylyl cyclase natriuretic peptide receptor-A in regulation of blood pressure and renal function. Physiol. Genom. 2018, 50, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.G. Human natriuretic peptide receptor—A guanylyl cyclase is self-associated prior to hormone binding. Biochemistry 1992, 31, 10421–10425. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L. Beyond blood pressure: The endothelium and atherosclerosis progression. Am. J. Hypertens. 2002, 15, 115s–122s. [Google Scholar] [CrossRef] [Green Version]
- Ichiki, T.; Izumi, R.; Cataliotti, A.; Larsen, A.M.; Sandberg, S.M.; Burnett, J.C., Jr. Endothelial permeability in vitro and in vivo: Protective actions of ANP and omapatrilat in experimental atherosclerosis. Peptides 2013, 48, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, C.; Liu, H.; Zhan, W.; Chen, L.; Yu, Z.; Tian, S.; Xiang, Y.; Chen, S.; Tian, X.L. Chronological attenuation of NPRA/PKG/AMPK signaling promotes vascular aging and elevates blood pressure. Aging Cell 2022, 21, e13699. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.J.; Lee, Y.J.; Kim, J.S.; Kang, D.G.; Lee, H.S. Protective role of betulinic acid on TNF-alpha-induced cell adhesion molecules in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 391, 96–101. [Google Scholar] [CrossRef]
- Jin, Y.P.; Nevarez-Mejia, J.; Terry, A.Q.; Sosa, R.A.; Heidt, S.; Valenzuela, N.M.; Rozengurt, E.; Reed, E.F. Cross-Talk between HLA Class I and TLR4 Mediates P-Selectin Surface Expression and Monocyte Capture to Human Endothelial Cells. J. Immunol. 2022, 209, 1359–1369. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Gogulamudi, V.R.; Mani, I.; Subramanian, U.; Pandey, K.N. Genetic disruption of Npr1 depletes regulatory T cells and provokes high levels of proinflammatory cytokines and fibrosis in the kidneys of female mutant mice. Am. J. Physiol. Physiol. 2019, 316, F1254–F1272. [Google Scholar] [CrossRef]
- Subramanian, U.; Kumar, P.; Mani, I.; Chen, D.; Kessler, I.; Periyasamy, R.; Raghavaraju, G.; Pandey, K.N. Retinoic acid and sodium butyrate suppress the cardiac expression of hypertrophic markers and proinflammatory mediators in Npr1 gene-disrupted haplotype mice. Physiol. Genom. 2016, 48, 477–490. [Google Scholar] [CrossRef]
- Rubattu, S.; Forte, M.; Marchitti, S.; Volpe, M. Molecular Implications of Natriuretic Peptides in the Protection from Hypertension and Target Organ Damage Development. Int. J. Mol. Sci. 2019, 20, 798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokudome, T.; Kishimoto, I.; Yamahara, K.; Osaki, T.; Minamino, N.; Horio, T.; Sawai, K.; Kawano, Y.; Miyazato, M.; Sata, M.; et al. Impaired recovery of blood flow after hind-limb ischemia in mice lacking guanylyl cyclase-A, a receptor for atrial and brain natriuretic peptides. Arter. Thromb. Vasc. Biol. 2009, 29, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Hurtubise, J.; McLellan, K.; Durr, K.; Onasanya, O.; Nwabuko, D.; Ndisang, J.F. The Different Facets of Dyslipidemia and Hypertension in Atherosclerosis. Curr. Atheroscler. Rep. 2016, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Cachofeiro, V.; Miana, M.; Heras, N.; Martín-Fernandez, B.; Ballesteros, S.; Balfagon, G.; Lahera, V. Inflammation: A Link Between Hypertension and Atherosclerosis. Curr. Hypertens. Rev. 2009, 5, 40–48. [Google Scholar] [CrossRef]
- Kevil, C.G.; Patel, R.P.; Bullard, D.C. Essential role of ICAM-1 in mediating monocyte adhesion to aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2001, 281, C1442–C1447. [Google Scholar] [CrossRef] [Green Version]
- Lawson, C.; Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 2009, 61, 22–32. [Google Scholar] [CrossRef]
- Ling, S.; Nheu, L.; Komesaroff, P.A. Cell adhesion molecules as pharmaceutical target in atherosclerosis. Mini. Rev. Med. Chem. 2012, 12, 175–183. [Google Scholar] [CrossRef]
- Ichiki, T.; Jougasaki, M.; Setoguchi, M.; Imamura, J.; Nakashima, H.; Matsuoka, T.; Sonoda, M.; Nakamura, K.; Minagoe, S.; Tei, C. Cardiotrophin-1 stimulates intercellular adhesion molecule-1 and monocyte chemoattractant protein-1 in human aortic endothelial cells. Am. J. Physiol. Circ. Physiol. 2008, 294, H750–H763. [Google Scholar] [CrossRef]
- Khodabandehlou, K.; Masehi-Lano, J.J.; Poon, C.; Wang, J.; Chung, E.J. Targeting cell adhesion molecules with nanoparticles using in vivo and flow-based in vitro models of atherosclerosis. Exp. Biol. Med. 2017, 242, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Potteaux, S.; Gautier, E.L.; Hutchison, S.B.; van Rooijen, N.; Rader, D.J.; Thomas, M.J.; Sorci-Thomas, M.G.; Randolph, G.J. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J. Clin. Investig. 2011, 121, 2025–2036. [Google Scholar] [CrossRef]
- Herbin, O.; Regelmann, A.G.; Ramkhelawon, B.; Weinstein, E.G.; Moore, K.J.; Alexandropoulos, K. Monocyte Adhesion and Plaque Recruitment During Atherosclerosis Development Is Regulated by the Adapter Protein Chat-H/SHEP1. Arter. Thromb. Vasc. Biol. 2016, 36, 1791–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, D. Airway epithelial integrins: Why so many? Am. J. Respir. Cell Mol. Biol. 1998, 19, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Elaimy, A.L.; Wang, M.; Sheel, A.; Brown, C.W.; Walker, M.R.; Amante, J.J.; Xue, W.; Chan, A.; Baer, C.E.; Goel, H.L.; et al. Real-time imaging of integrin β4 dynamics using a reporter cell line generated by Crispr/Cas9 genome editing. J. Cell Sci. 2019, 132, jcs231241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.Y.; Araki, S.; Kaji, K.; Hayashi, H. Integrin beta4 is involved in apoptotic signal transduction in endothelial cells. Biochem. Biophys. Res. Commun. 1997, 233, 182–186. [Google Scholar] [CrossRef]
- Homan, S.M.; Mercurio, A.M.; LaFlamme, S.E. Endothelial cells assemble two distinct alpha6beta4-containing vimentin-associated structures: Roles for ligand binding and the beta4 cytoplasmic tail. J. Cell Sci. 1998, 111 Pt 18, 2717–2728. [Google Scholar] [CrossRef]
- Frijns, E.; Sachs, N.; Kreft, M.; Wilhelmsen, K.; Sonnenberg, A. EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin β4. J. Biol. Chem. 2010, 285, 37650–37662. [Google Scholar] [CrossRef] [Green Version]
- Kariya, Y.; Gu, J. N-glycosylation of ß4 integrin controls the adhesion and motility of keratinocytes. PLoS ONE 2011, 6, e27084. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Epshtein, Y.; Ni, X.; Dull, R.O.; Cress, A.E.; Garcia, J.G.; Jacobson, J.R. Role of Integrin β4 in Lung Endothelial Cell Inflammatory Responses to Mechanical Stress. Sci. Rep. 2015, 5, 16529. [Google Scholar] [CrossRef] [Green Version]
- De Vito, P. Atrial natriuretic peptide: An old hormone or a new cytokine? Peptides 2014, 58, 108–116. [Google Scholar] [CrossRef]
- Long, C.; Liu, H.; Zhan, W.; Chen, L.; Wu, A.; Yang, L.; Chen, S. Null Function of Npr1 Disturbs Immune Response in Colonic Inflammation during Early Postnatal Stage. Inflammation 2022. published online ahead of print. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Liu, J.; Long, C.; Chen, L.; Zhan, W.; Xiao, W.; Gong, X.; Liu, M.; Tian, X.-L.; Chen, S. Lack of NPR1 Increases Vascular Endothelial Adhesion through Induction of Integrin Beta 4. Int. J. Mol. Sci. 2022, 23, 12627. https://doi.org/10.3390/ijms232012627
Liu H, Liu J, Long C, Chen L, Zhan W, Xiao W, Gong X, Liu M, Tian X-L, Chen S. Lack of NPR1 Increases Vascular Endothelial Adhesion through Induction of Integrin Beta 4. International Journal of Molecular Sciences. 2022; 23(20):12627. https://doi.org/10.3390/ijms232012627
Chicago/Turabian StyleLiu, Hongfei, Jiankun Liu, Changkun Long, Liping Chen, Wenxing Zhan, Wanli Xiao, Xueting Gong, Man Liu, Xiao-Li Tian, and Shenghan Chen. 2022. "Lack of NPR1 Increases Vascular Endothelial Adhesion through Induction of Integrin Beta 4" International Journal of Molecular Sciences 23, no. 20: 12627. https://doi.org/10.3390/ijms232012627
APA StyleLiu, H., Liu, J., Long, C., Chen, L., Zhan, W., Xiao, W., Gong, X., Liu, M., Tian, X.-L., & Chen, S. (2022). Lack of NPR1 Increases Vascular Endothelial Adhesion through Induction of Integrin Beta 4. International Journal of Molecular Sciences, 23(20), 12627. https://doi.org/10.3390/ijms232012627