

Development of Anti-LRRC15 Small Fragments for Imaging Purposes Using a Phage-Display ScFv Approach
 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Immunization

2.2. Library Construction
- -
- The size, assessed by spotting 5 µL of 10 times serial dilution of TG1 cells after electroporation. For the given testing conditions, an acceptable size was >106 colony-forming unit (cfu/µg).
- -
- The insertion rate (IR), determined after colony PCR on randomly chosen clones from the bacterial spread of the electroporated library. A clone was deemed positive when the PCR indicated a 1100 bp fragment. IR was assessed as acceptable when it included more than 80% of positive clones.
- -
- The quality of PCR inserts cloned in our phagemid (expression vector usable for phage display) was also assessed by the sequencing of 96 randomly chosen positive PCR clones, to determine the % of coding ScFv sequences showing an ORF. In our study, the number of sequences presenting an ScFv ORF was >90%, which was higher than our own criteria of >80% (Table 2). To validate the library, the global diversity was confirmed by a low level of identical sequences (less than 10%). In this study, no redundance was found for the analyzed clones. Summaries of clone sequences and diversity analyses are presented in Supplementary Data S3.
2.3. Panning—Selection
2.4. Screening by Phage Flow Cytometry
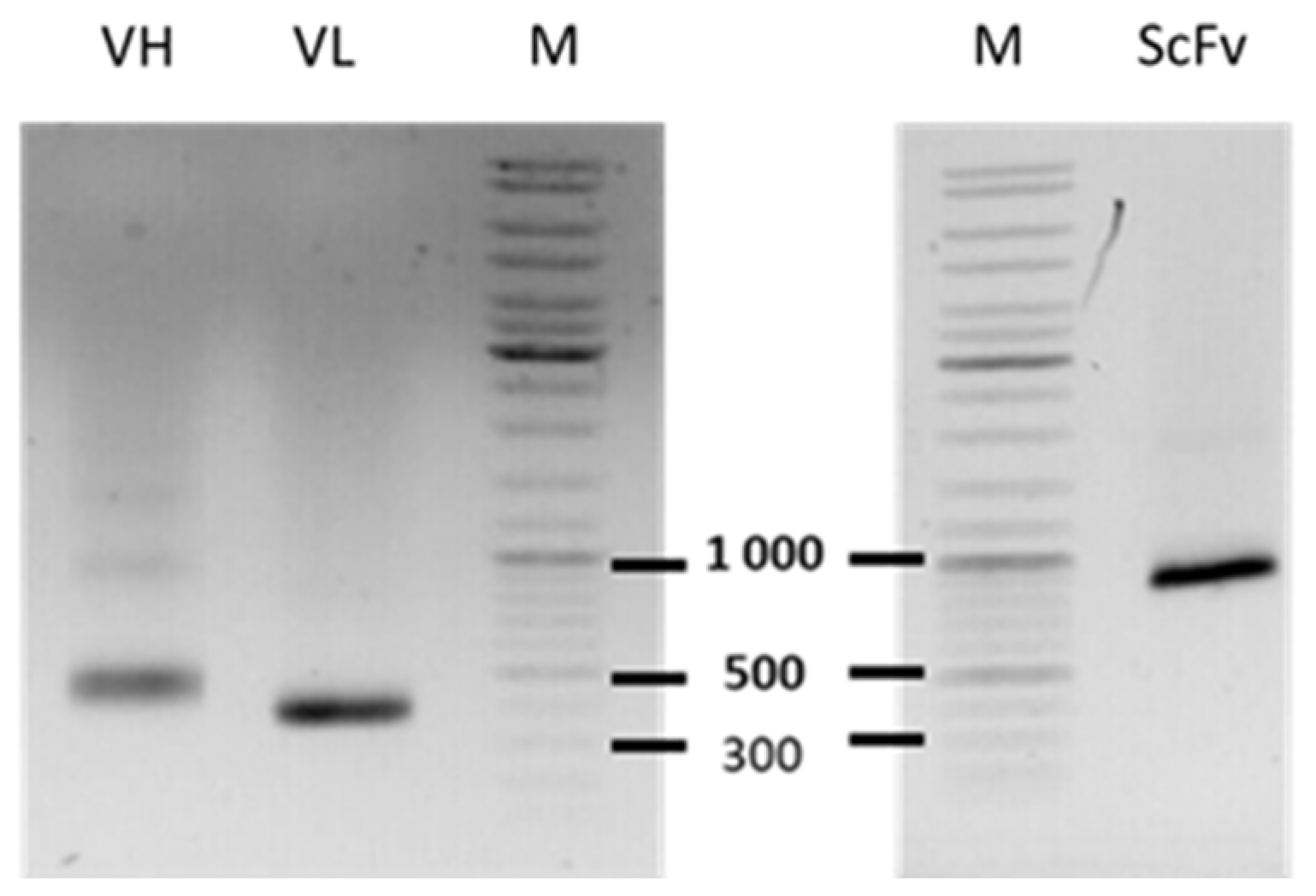
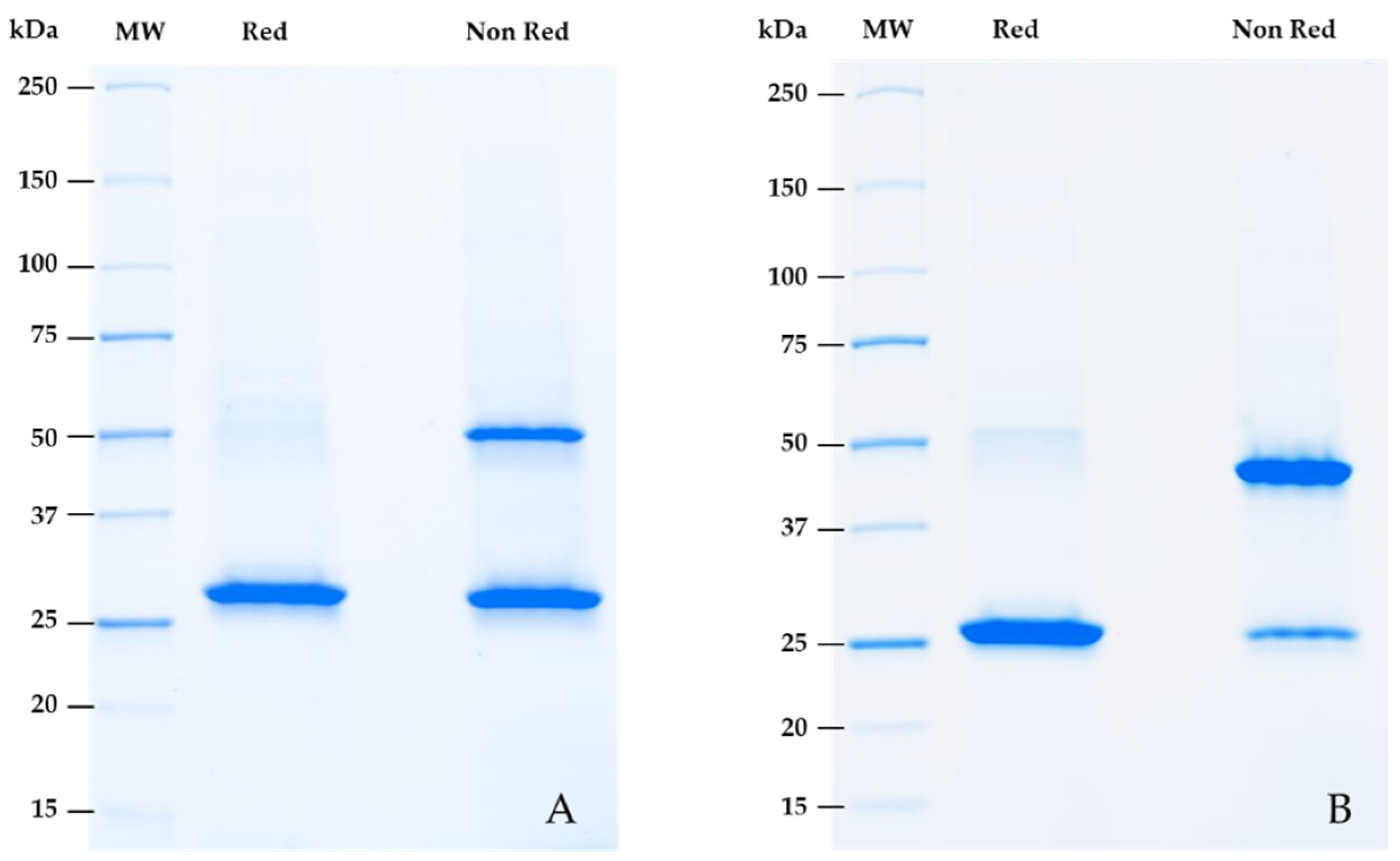
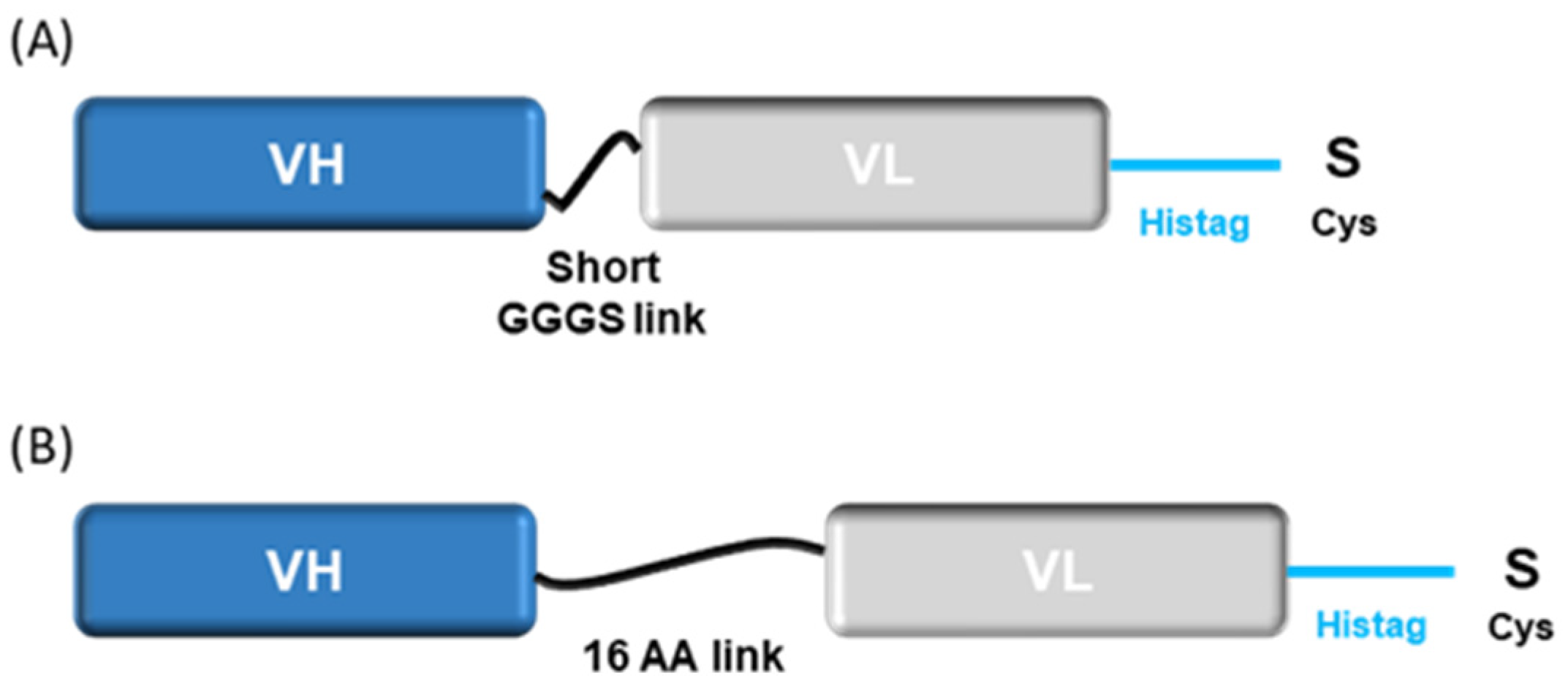
2.5. Reformatting and Validation of Small Recombinant Fragments
2.6. Validation of Small Engineering Recombinant Fragments
3. Discussion
4. Materials and Methods
4.1. Human LRRC15
4.1.1. Antigen for Immunization
4.1.2. Cell Lines
4.2. Murine LRRC15
4.3. Reagents
4.4. Immunization
4.5. ScFv Library
4.5.1. Construction
4.5.2. Validation
4.5.3. Phage Infection and preparation
4.6. Phage Display
4.6.1. Panning-Selection
4.6.2. Screening
4.7. Engineering of Candidates
4.7.1. Design and Cloning
4.7.2. Validation of Final Products
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The International Agency for Research on Cancer. Available online: https://gco.iarc.fr/today/home (accessed on 14 June 2022).
- Park, I.; Kwon, M.; Shin, E.-C. Immune checkpoint inhibitors for cancer treatment. Arch. Pharm. Res. 2016, 39, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Kourie, H.R.; Awada, G.; Awada, A.H. Learning from the “tsunami” of immune checkpoint inhibitors in 2015. Crit. Rev. Oncol. Hematol. 2016, 101, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Kesmir, C.; van Buuren, M.M. Biomarkers in cancer immunotherapy. Cancer Cell 2015, 27, 12–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–285. [Google Scholar] [CrossRef]
- Tan, W.C.C.; Nerurkar, S.A.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/ immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [Green Version]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2002, 11, 725–732. [Google Scholar] [CrossRef]
- Satoh, K.; Hata, M.; Yokota, H. A novel member of the leucine-rich repeat superfamily induced in rat astrocytes by β-amyloid. Biochem. Biophys. Res. Commun. 2002, 290, 756–762. [Google Scholar] [CrossRef]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased Expression of Genes Converting Adrenal Androgens to Testosterone in Androgen-Independent Prostate Cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef]
- Satoh, K.; Hata, M.; Yokota, H. High Lib mRNA expression in breast carcinomas. DNA Res. 2004, 11, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Prey, J.; Wilkinson, S.; Ryan, K.M. Tumor Antigen LRRC15 Impedes Adenoviral Infection: Implications for Virus-Based Cancer Therapy. J. Virol. 2008, 82, 5933–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, J.W.; Tanlimco, S.G.; Hickson, J.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; Mcgonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody-Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15 + Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Liu, W.; Xu, Z.; Zhao, J.; Wang, W.; Yu, Z.; Wei, M. Integrated microenvironment-associated genomic profiles identify LRRC15 mediating recurrent glioblastoma-associated macrophages infiltration. J. Cell. Mol. Med. 2021, 25, 5534–5546. [Google Scholar] [CrossRef]
- Ray, U.; Jung, D.-B.; Jin, L.; Xiao, Y.; Dasari, S.; Sarkar Bhattacharya, S.; Thirusangu, P.; Staub, J.K.; Roy, D.; Roy, B.; et al. Targeting LRRC15 Inhibits Metastatic Dissemination of Ovarian Cancer. Cancer Res. 2022, 82, 1038–1054. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Perret, R.; Huang, Y.; Courgeon, F.; Gokhale, P.C.; Laroche-Clary, A.; Eschle, B.K.; Velasco, V.; Le Loarer, F.; Algeo, M.P.; et al. LRRC15 Targeting in Soft-Tissue Sarcomas: Biological and Clinical Implications. Cancers 2020, 12, 757. [Google Scholar] [CrossRef] [Green Version]
- Slemmons, K.K.; Mukherjee, S.; Meltzer, P.; Purcell, J.W.; Helman, L.J. LRRC15 antibody-drug conjugates show promise as osteosarcoma therapeutics in preclinical studies. Pediatr. Blood Cancer 2021, 68, e28771. [Google Scholar] [CrossRef]
- Demetri, G.D.; Luke, J.J.; Hollebecque, A.; Powderly, J.D., II; Spira, A.I.; Subbiah, V.; Naumovski, L.; Chen, C.; Fang, H.; Lai, D.W.; et al. First-in-Human Phase I Study of ABBV-085, an Antibody-Drug Conjugate Targeting LRRC15, in Sarcomas and Other Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3556–3566. [Google Scholar] [CrossRef]
- Ray, U.; Pathoulas, C.L.; Thirusangu, P.; Purcell, J.W.; Kannan, N.; Shridhar, V. Exploiting LRRC15 as a Novel Therapeutic Target in Cancer. Cancer Res. 2022, 82, 1675–1681. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olafsen, T.; Sirk, S.; Olma, S.; Shen, K.F.; Wu, A. ImmunoPET using engineered antibody fragments: Fluorine-18 labeled diabodies for same-day imaging. Tumour. Biol. 2012, 33, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Reader, R.H.; Workman, R.G.; Maddison, B.C.; Gough, K.C. Advances in the Production and Batch Reformatting of Phage Antibody Libraries. Mol. Biotechnol. 2019, 61, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Olafsen, T.; Cheung, C.; Yazaki, P.; Li, L.; Sundaresan, G.; Gambhir, S.; Sherman, M.; Williams, L.; Shively, J.; Raubitschek, A.; et al. Covalent disulfide-linked anti-CEA diabody allows site-specific conjugation and radiolabeling for tumor targeting applications. Protein Eng. Des. Sel. 2004, 17, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Sirk, S.J.; Olafsen, T.; Barat, B.; Bauer, K.B.; Wu, A.M. Site-specific, thiol-mediated conjugation of fluorescent probes to cysteine-modified diabodies targeting CD20 or HER2. Bioconjug. Chem. 2008, 19, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.; Carroll, L.; Yahioglu, G.; Aboagye, E.; Miller, P. Antibody Fragment and Affibody ImmunoPET Imaging Agents: Radiolabelling Strategies and Applications. ChemMedChem 2018, 13, 2466–2478. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Nian, S.; Li, L.; Wen, X.; Liu, Q.; Zhang, B.; Lan, Y.; Yuan, Q.; Ye, Y. Fully human recombinant antibodies against EphA2 from a multi-tumor patient immune library suitable for tumor-targeted therapy. Bioengineered 2021, 12, 10379–10400. [Google Scholar] [CrossRef]
- Duan, S.; Jia, Y.; Xie, D.; Xiao, S.; Zhou, C.; Zeng, F. Selection of novel human scFvs against cancer antigen IL1RAP by phage and yeast surface display technology. Biotechnol. Biotechnol. Equip. 2020, 34, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Sakemura, R.; Can, I.; Siegler, E.; Kenderian, S. In vivo CART cell imaging: Paving the way for success in CART cell therapy. Mol. Ther. Oncolytics 2021, 20, 625–633. [Google Scholar] [CrossRef]
- Kellner, C.; Nodehi, S.M.; Peipp, M. Mouse Immune Libraries for the Generation of ScFv Fragments Directed against Human Cell Surface Antigens; Kontermann, R., Dübel, S., Eds.; Antibody Engineering, Springer Protocols Handbooks; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar] [CrossRef]
- Russo, G.; Meier, D.; Helmsing, S.; Wenzel, E.; Oberle, F.; Frenzel, A.; Hust, M. Parallelized Antibody Selection in Microtiter Plates. Methods Mol. Biol. 2018, 1701, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Schirrmann, T. Binding Studies with Flow Cytometry; Kontermann, R., Dübel, S., Eds.; Antibody Engineering. Springer Protocols Handbooks; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Cells × 106 | M1 | M2 | M3 | M4 | M5 | M6 | Total |
|---|---|---|---|---|---|---|---|
| Lymph nodes | 46 | 74 | 86 | 132 | 98 | 114 | 550 |
| Used for phage display | 30 | 30 | 30 | 30 | 30 | 30 | 180 |
| Final ScFv Library | ||||
|---|---|---|---|---|
| Size (cfu/µg) | 1.2 × 107 | |||
| Insertion rate (colony PCR) | >95% (151/156 positives) | |||
| Sequencing control (96 clones) | >90% coding sequences (88/96)
100% of diversity (88/88) | |||
| Panning | Cells | Input (pfu) | Output (pfu) | Ratio (Out/In) |
| 1st round | 5 × 106 HeLa hLRRC15 | 1 × 1011 | 3 × 104 | 3 × 10−7 |
| 2nd round | 5 × 106 NIH3T3 hLRRC15 | 1 × 108 | 2 × 105 | 2 × 10−3 |
| Screen | Form | Cells | Candidate Numbers |
|---|---|---|---|
| 1 | Recombinant human | HeLa and NIH3T3 hLRRC15 | 80/90 |
| 2 | Natural human | U87-MG + TGFb | 28/80 |
| 3 | Recombinant murine | HEK 293 mLRRC15 | 2/28 |
| Sequence Number | Duplicate Clones | Family (CDR3) | |
|---|---|---|---|
| VH | 28 | 3 | 17 |
| VL | 28 | 3 | 24 |
| Concentration (mg/mL) | Quantity (mg) | |
|---|---|---|
| F4 Cys-ScFv | 1 | 4.5 |
| F4 Cys-Db | 1 | 1 |
| B3.1 Cys-ScFv | 1 | 1.3 |
| B3.1 Cys-Db | 1 | 2.9 |
| Human Natural | Human Recombinant | Murine Recombinant | ||
|---|---|---|---|---|
| % LRRC15 | U87-MG
(0.125 µg/test) | HeLa
(0.125 µg/test) | NIH3T3
(0.125 µg/test) | HEK 293
(0.125 µg/test) |
| F4 ScFv | 96.4 | 72.4 | 64 | 44.1 |
| B3.1 ScFv | 37.9 | 41.5 | 11 | 18.9 |
| F4 Db | 37.7 | 51.2 | 51 | 56.3 |
| B3.1 Db | 12.3 | 20.7 | 37 | 41.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baurand, P.-E.; Balland, J.; Reynas, C.; Ramseyer, M.; Vivier, D.; Bellaye, P.-S.; Collin, B.; Paul, C.; Denat, F.; Asgarov, K.; et al. Development of Anti-LRRC15 Small Fragments for Imaging Purposes Using a Phage-Display ScFv Approach. Int. J. Mol. Sci. 2022, 23, 12677. https://doi.org/10.3390/ijms232012677
Baurand P-E, Balland J, Reynas C, Ramseyer M, Vivier D, Bellaye P-S, Collin B, Paul C, Denat F, Asgarov K, et al. Development of Anti-LRRC15 Small Fragments for Imaging Purposes Using a Phage-Display ScFv Approach. International Journal of Molecular Sciences. 2022; 23(20):12677. https://doi.org/10.3390/ijms232012677
Chicago/Turabian StyleBaurand, Pierre-Emmanuel, Jérémy Balland, Chloé Reynas, Mélanie Ramseyer, Delphine Vivier, Pierre-Simon Bellaye, Bertrand Collin, Catherine Paul, Franck Denat, Kamal Asgarov, and et al. 2022. "Development of Anti-LRRC15 Small Fragments for Imaging Purposes Using a Phage-Display ScFv Approach" International Journal of Molecular Sciences 23, no. 20: 12677. https://doi.org/10.3390/ijms232012677
APA StyleBaurand, P. -E., Balland, J., Reynas, C., Ramseyer, M., Vivier, D., Bellaye, P. -S., Collin, B., Paul, C., Denat, F., Asgarov, K., Pallandre, J. -R., & Ringenbach, L. (2022). Development of Anti-LRRC15 Small Fragments for Imaging Purposes Using a Phage-Display ScFv Approach. International Journal of Molecular Sciences, 23(20), 12677. https://doi.org/10.3390/ijms232012677