
Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications

Abstract
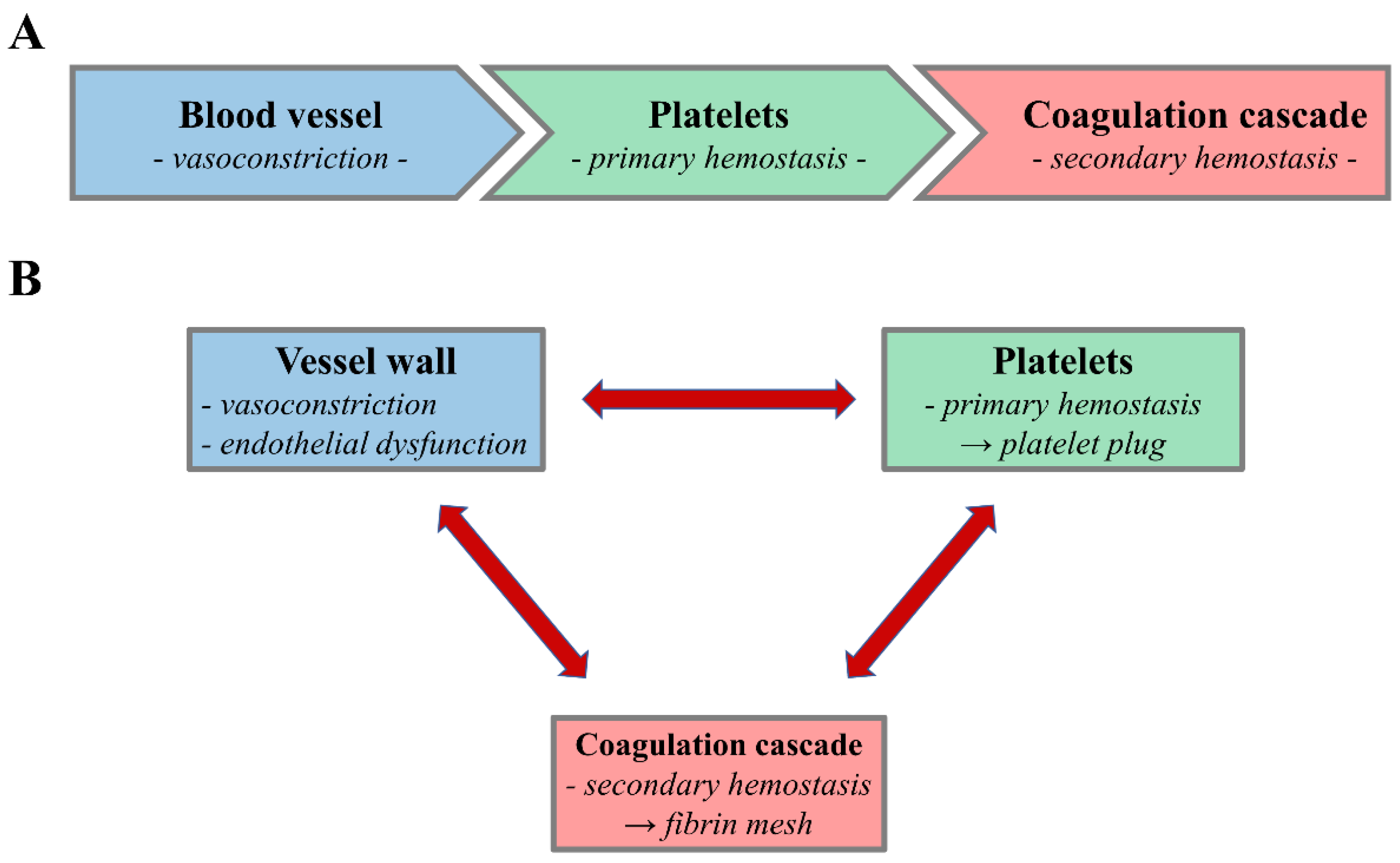
:1. Introduction
2. Platelet Shape and Structure at Rest and during Activation
3. Role of Platelets in (Primary) Hemostasis
4. Clinical Implications of Platelet (Patho)Physiology
4.1. Platelet Abnormalities Translate into Clinically Relevant Dysfunctions of (Not Only) Primary Hemostasis
4.2. The Functional Properties of Platelets Explain Their Stronger Implications in Arterial Than Venous Thromboses
4.3. Antiplatelet (and Anticoagulant) Drugs Target Key Mechanisms of Platelets’ Adhesion, Activation, and Aggregation
5. Gaps in Knowledge and Future Research
6. Conclusions
Funding
Conflicts of Interest
References
- Rumbaut, R.E.; Thiagarajan, P. Chapter 2, General Characteristics of Platelets. In Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Rondina, M.T.; Zimmerman, G.A. The Role of Platelets in Inflammation in Platelets, 4th ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 505–522. [Google Scholar] [CrossRef]
- Alsousou, J.; Harrison, P. Therapeutic Platelet-Rich Plasma in Wound Healing in Platelets, 4th ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 1161–1171. [Google Scholar] [CrossRef]
- Horowitz, A.M.; Villeda, S.A. Therapeutic potential of systemic brain rejuvenation strategies for neurodegenerative disease. F1000Research 2017, 6, 1291. [Google Scholar] [CrossRef] [Green Version]
- Aquino-Domínguez, A.S.; Romero-Tlalolini, M.L.A.; Torres-Aguilar, H.; Aguilar-Ruiz, S.R. Recent advances in the discovery and function of antimicrobial molecules in platelets. Int. J. Mol. Sci. 2021, 22, 10230. [Google Scholar] [CrossRef]
- Yeaman, M.R. The Role of Platelets in Antimicrobial Host Defense in Platelets, 4th ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 523–546. [Google Scholar] [CrossRef]
- Eriksson, O.; Mohlin, C.; Nilsson, B.; Ekdahl, K.N. The human platelet as an innate immune cell: Interactions between activated platelets and the complement System. Front. Immunol. 2019, 10, 1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coller, B.S. Biochemical and electrostatic considerations in primary platelet aggregation. Ann. N. Y. Acad. Sci. 1983, 416, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Sage, S.O.; Pugh, N.; Farndale, R.W.; Harper, A.G. Pericellular Ca(2+) recycling potentiates thrombin-evoked Ca(2+) signals in human platelets. Physiol. Rep. 2013, 1, e00085. [Google Scholar] [CrossRef]
- Grozovsky, R.; Begonja, A.J.; Liu, K.; Visner, G.; Hartwig, J.H.; Falet, H.; Hoffmeister, K.M. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat. Med. 2015, 21, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.G. The Structure of Resting and Activated Platelets in Platelets, 4th ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 47–77. [Google Scholar] [CrossRef]
- Coughlin, S.R. Protease-activated receptors in vascular biology. Thromb. Haemost. 2001, 86, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, H.; Zeng, D.; Connolly, A.J.; Tam, C.; Coughlin, S.R. Antibodies to protease-activated receptor 3 inhibit activation of mouse platelets by thrombin. Blood 1998, 91, 4152–4157. [Google Scholar] [CrossRef]
- Price, M.J. Bedside evaluation of thienopyridine antiplatelet therapy. Circulation 2009, 119, 2625–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, J.M.; Gambaryan, S.; Watson, S.P.; Jurk, K.; Walter, U.; Sickmann, A.; Heemskerk, J.W.; Zahedi, R.P. What can proteomics tell us about platelets? Circ. Res. 2014, 114, 1204–1219. [Google Scholar] [CrossRef] [Green Version]
- Kamykowski, J.; Carlton, P.; Sehgal, S.; Storrie, B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet α-granules. Blood 2011, 118, 1370–1373. [Google Scholar] [CrossRef] [Green Version]
- Bombeli, T.; Schwartz, B.R.; Harlan, J.M. Adhesion of activated platelets to endothelial cells: Evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J. Exp. Med. 1998, 187, 329–339. [Google Scholar] [CrossRef]
- Guidetti, G.F.; Bernardi, B.; Consonni, A.; Rizzo, P.; Gruppi, C.; Balduini, C.; Torti, M. Integrin alpha2beta1 induces phosphorylation-dependent and phosphorylation-independent activation of phospholipase Cgamma2 in platelets: Role of Src kinase and Rac GTPase. J. Thromb. Haemost. 2009, 7, 1200–1206. [Google Scholar] [CrossRef]
- Rosado, J.A.; Sage, S.O. A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Trends Cardiovasc. Med. 2000, 10, 327–332. [Google Scholar] [CrossRef]
- Nakahata, N. Thromboxane A2: Physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol. Ther. 2008, 118, 18–35. [Google Scholar] [CrossRef]
- Lee, R.H.; Stefanini, L.; Bergmeier, W. Platelet Signal Transduction in Platelets, 4th ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 329–348. [Google Scholar] [CrossRef]
- Fadeel, B.; Xue, D. The ins and outs of phospholipid asymmetry in the plasma membrane: Roles in health and disease. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Schoenwaelder, S.M. Procoagulant platelets: Are they necrotic? Blood 2010, 116, 2011–2018. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, J.W.; Bevers, E.M.; Lindhout, T. Platelet activation and blood coagulation. Thromb. Haemost. 2002, 88, 186–193. [Google Scholar] [PubMed]
- Bledzka, K.; Qin, J.; Plow, E.F. Integrin αIIbβ3 in Platelets, 4th ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 227–241. [Google Scholar] [CrossRef]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef]
- Quek, L.S.; Pasquet, J.M.; Hers, I.; Cornall, R.; Knight, G.; Barnes, M.; Hibbs, M.L.; Dunn, A.R.; Lowell, C.A.; Watson, S.P. Fyn and Lyn phosphorylate the Fc receptor gamma chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood 2000, 96, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasne, D.; Jude, B.; Susen, S. From normal to pathological hemostasis. Can. J. Anaesth. 2006, 53 (Suppl. 6), S2–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camera, M.; Brambilla, M.; Toschi, V.; Tremoli, E. Tissue factor expression on platelets is a dynamic event. Blood 2010, 116, 5076–5077. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, L.M.; Liu, M.L. Microvesicles and diabetic complications--novel mediators, potential biomarkers and therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Duckers, C.; Simioni, P.; Spiezia, L.; Radu, C.; Dabrilli, P.; Gavasso, S.; Rosing, J.; Castoldi, E. Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood 2010, 115, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Bowie, E.J.; Solberg, L.A., Jr.; Fass, D.N.; Johnson, C.M.; Knutson, G.J.; Stewart, M.L.; Zoecklein, L.J. Transplantation of normal bone marrow into a pig with severe von Willebrand’s disease. J. Clin. Investig. 1986, 78, 26–30. [Google Scholar] [CrossRef]
- Arthur, J.F.; Gardiner, E.E.; Matzaris, M.; Taylor, S.G.; Wijeyewickrema, L.; Ozaki, Y.; Kahn, M.L.; Andrews, R.K.; Berndt, M.C. Glycoprotein VI is associated with GPIb-IX-V on the membrane of resting and activated platelets. Thromb. Haemost. 2005, 93, 716–723. [Google Scholar] [CrossRef]
- Szanto, T.; Nummi, V.; Jouppila, A.; Brinkman, H.J.M.; Lassila, R. Platelets compensate for poor thrombin generation in type 3 von Willebrand disease. Platelets 2020, 31, 103–111. [Google Scholar] [CrossRef]
- Koike, Y.; Tanaka, K.; Okugawa, Y.; Morimoto, Y.; Toiyama, Y.; Uchida, K.; Miki, C.; Mizoguchi, A.; Kusunoki, M. In vivo real-time two-photon microscopic imaging of platelet aggregation induced by selective laser irradiation to the endothelium created in the beta-actin-green fluorescent protein transgenic mice. J. Thromb. Thrombolysis. 2011, 32, 138–145. [Google Scholar] [CrossRef]
- Konstantinides, S.V.; Meyer, G.; Becattini, C.; Bueno, H.; Geersing, G.J.; Harjola, V.P.; Huisman, M.V.; Humbert, M.; Jennings, C.S.; Jiménez, D.; et al. 2019 ESC Guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS). Eur. Heart J. 2020, 41, 543–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valgimigli, M.; Bueno, H.; Byrne, R.A.; Collet, J.P.; Costa, F.; Jeppsson, A.; Jüni, P.; Kastrati, A.; Kolh, P.; Mauri, L.; et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: The Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2018, 39, 213–260. [Google Scholar] [CrossRef] [PubMed]
- Onasoga-Jarvis, A.A.; Puls, T.J.; O’Brien, S.K.; Kuang, L.; Liang, H.J.; Neeves, K.B. Thrombin generation and fibrin formation under flow on biomimetic tissue factor-rich surfaces. J. Thromb. Haemost. 2014, 12, 373–382. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Heresi, G.A.; Velasquez, H.; Jy, W.; Jimenez, J.J.; Ahn, E.; Horstman, L.L.; Soriano, A.O.; Zambrano, J.P.; Ahn, Y.S. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J. Am. Coll. Cardiol. 2005, 45, 1467–1471. [Google Scholar] [CrossRef]
- von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Tarantino, E.; Amadio, P.; Squellerio, I.; Porro, B.; Sandrini, L.; Turnu, L.; Cavalca, V.; Tremoli, E.; Barbieri, S.S. Role of thromboxane-dependent platelet activation in venous thrombosis: Aspirin effects in mouse model. Pharmacol. Res. 2016, 107, 415–425. [Google Scholar] [CrossRef]
- Mistry, D.A.; Chandratreya, A.; Lee, P.Y.F. A Systematic Review on the Use of Aspirin in the Prevention of Deep Vein Thrombosis in Major Elective Lower Limb Orthopedic Surgery: An Update from the Past 3 Years. Surg. J. 2017, 3, e191–e196. [Google Scholar] [CrossRef] [Green Version]
- Simes, J.; Becattini, C.; Agnelli, G.; Eikelboom, J.W.; Kirby, A.C.; Mister, R.; Prandoni, P.; Brighton, T.A.; INSPIRE Study Investigators (International Collaboration of Aspirin Trials for Recurrent Venous Thromboembolism). Aspirin for the prevention of recurrent venous thromboembolism: The INSPIRE collaboration. Circulation 2014, 130, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Lensing, A.W.A.; Prins, M.H.; Bauersachs, R.; Beyer-Westendorf, J.; Bounameaux, H.; Brighton, T.A.; Cohen, A.T.; Davidson, B.L.; Decousus, H.; et al. Rivaroxaban or Aspirin for Extended Treatment of Venous Thromboembolism. N. Engl. J. Med. 2017, 376, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly, S.; Pogue, J.; Hart, R.; Pfeffer, M.; Hohnloser, S.; Chrolavicius, S.; Pfeffer, M.; Hohnloser, S.; Yusuf, S. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): A randomised controlled trial. Lancet 2006, 367, 1903–1912. [Google Scholar] [CrossRef]
- Connolly, S.J.; Eikelboom, J.; Joyner, C.; Diener, H.C.; Hart, R.; Golitsyn, S.; Flaker, G.; Avezum, A.; Hohnloser, S.H.; Diaz, R.; et al. Apixaban in patients with atrial fibrillation. N. Engl. J. Med. 2011, 364, 806–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. Comparative Effectiveness of Pulmonary Embolism Prevention after Hip and Knee Replacement (PEPPER). Available online: https://clinicaltrials.gov/ct2/show/NCT02810704 (accessed on 16 August 2022).
- Koenen, R.R.; Binder, C.J. Platelets and coagulation factors: Established and novel roles in atherosclerosis and atherothrombosis. Atherosclerosis 2020, 307, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without Aspirin in Stable Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.J.; Gibson, C.M.; Yee, M.K.; Korjian, S.; Daaboul, Y.; Plotnikov, A.N.; Burton, P.; Braunwald, E. Safety and Efficacy of Rivaroxaban When Added to Aspirin Monotherapy Among Stabilized Post-Acute Coronary Syndrome Patients: A Pooled Analysis Study of ATLAS ACS-TIMI 46 and ATLAS ACS 2-TIMI 51. J. Am. Heart Assoc. 2019, 8, e009451. [Google Scholar] [CrossRef]
- Effect of long-term oral anticoagulant treatment on mortality and cardiovascular morbidity after myocardial infarction. Anticoagulants in the Secondary Prevention of Events in Coronary Thrombosis (ASPECT) Research Group. Lancet 1994, 343, 499–503.
- Nehaj, F.; Sokol, J.; Ivankova, J.; Mokan, M.; Mokan, M.; Stasko, J. Edoxaban affects TRAP-dependent platelet aggregation. J. Thromb. Thrombolysis 2020, 49, 578–583. [Google Scholar] [CrossRef]
- Eller, T.; Busse, J.; Dittrich, M.; Flieder, T.; Alban, S.; Knabbe, C.; Birschmann, I. Dabigatran, rivaroxaban, apixaban, argatroban and fondaparinux and their effects on coagulation POC and platelet function tests. Clin. Chem. Lab. Med. 2014, 52, 835–844. [Google Scholar] [CrossRef]
- Tsantes, A.E.; Kyriakou, E.; Bonovas, S.; Chondrogianni, M.; Zompola, C.; Liantinioti, C.; Simitsi, A.; Katsanos, A.H.; Atta, M.; Ikonomidis, I.; et al. Impact of dabigatran on platelet function and fibrinolysis. J. Neurol. Sci. 2015, 357, 204–208. [Google Scholar] [CrossRef]
- Kyriakou, E.; Ikonomidis, I.; Stylos, D.; Bonovas, S.; Papadakis, I.; Nikolopoulos, G.K.; Kokoris, S.; Kalantzis, D.; Economopoulou, C.; Kopterides, P.; et al. Laboratory assessment of the anticoagulant activity of dabigatran. Clin. Appl. Thromb. Hemost. 2015, 21, 434–445. [Google Scholar] [CrossRef]
- Artang, R.; Rome, E.; Nielsen, J.D.; Vidaillet, H.J. Meta-analysis of randomized controlled trials on risk of myocardial infarction from the use of oral direct thrombin inhibitors. Am. J. Cardiol. 2013, 112, 1973–1979. [Google Scholar] [CrossRef] [Green Version]
- López-López, J.A.; Sterne, J.A.C.; Thom, H.H.Z.; Higgins, J.P.T.; Hingorani, A.D.; Okoli, G.N.; Davies, P.A.; Bodalia, P.N.; Bryden, P.A.; Welton, N.J.; et al. Oral anticoagulants for prevention of stroke in atrial fibrillation: Systematic review, network meta-analysis, and cost effectiveness analysis. BMJ 2017, 359, j5058, Erratum in BMJ 2017, 359, j5631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petzold, T.; Thienel, M.; Konrad, I.; Schubert, I.; Regenauer, R.; Hoppe, B.; Lorenz, M.; Eckart, A.; Chandraratne, S.; Lennerz, C.; et al. Oral thrombin inhibitor aggravates platelet adhesion and aggregation during arterial thrombosis. Sci. Transl. Med. 2016, 8, 367ra168. [Google Scholar] [CrossRef] [PubMed]
- Achilles, A.; Mohring, A.; Dannenberg, L.; Grandoch, M.; Hohlfeld, T.; Fischer, J.W.; Levkau, B.; Kelm, M.; Zeus, T.; Polzin, A. Dabigatran enhances platelet reactivity and platelet thrombin receptor expression in patients with atrial fibrillation. J. Thromb. Haemost. 2017, 15, 473–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scridon, A.; Mărginean, A.; Huțanu, A.; Chinezu, L.; Gheban, D.; Perian, M.; Vântu, A.; Gherțescu, D.; Fișcă, P.C.; Șerban, R.C.; et al. Vascular protease-activated receptor 4 upregulation, increased platelet aggregation, and coronary lipid deposits induced by long-term dabigatran administration—results from a diabetes animal model. J. Thromb. Haemost. 2019, 17, 538–550. [Google Scholar] [CrossRef]
- Olivier, C.B.; Weik, P.; Meyer, M.; Weber, S.; Anto-Michel, N.; Diehl, P.; Zhou, Q.; Geisen, U.; Bode, C.; Moser, M. TRAP-induced platelet aggregation is enhanced in cardiovascular patients receiving dabigatran. Thromb. Res. 2016, 138, 63–68. [Google Scholar] [CrossRef]
- Han, X.; Nieman, M.T.; Kerlin, B.A. Protease-activated receptors: An illustrated review. Res. Pract. Thromb. Haemost. 2020, 5, 17–26. [Google Scholar] [CrossRef]
- Fareed, J.; Hoppensteadt, D.; Maddenini, J.; Schultz, J.; Neville, B.; Iqbal, O.; Perzborn, E.; Misselwitz, F. Antithrombotic mechanism of action of BAY 59-7939—A novel, oral, direct factor Xa inhibitor. J. Thromb. Haemost. 2005, 3, P0518. [Google Scholar]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Misselwitz, F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discov. 2011, 10, 61–75. [Google Scholar] [CrossRef]
- Steppich, B.; Dobler, F.; Brendel, L.C.; Hessling, G.; Braun, S.L.; Steinsiek, A.L.; Deisenhofer, I.; Hyseni, A.; Roest, M.; Ott, I. Effect of the FXa inhibitors Rivaroxaban and Apixaban on platelet activation in patients with atrial fibrillation. J. Thromb. Thrombolysis 2017, 43, 490–497. [Google Scholar] [CrossRef]
- Perzborn, E.; Heitmeier, S.; Laux, V. Effects of Rivaroxaban on Platelet Activation and Platelet-Coagulation Pathway Interaction: In Vitro and In Vivo Studies. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Pujadas-Mestres, L.; Lopez-Vilchez, I.; Arellano-Rodrigo, E.; Reverter, J.C.; Lopez-Farre, A.; Diaz-Ricart, M.; Badimon, J.J.; Escolar, G. Differential inhibitory action of apixaban on platelet and fibrin components of forming thrombi: Studies with circulating blood and in a platelet-based model of thrombin generation. PLoS ONE 2017, 12, e0171486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahito, K.; Adachi, H.; Ino, T. Platelet aggregation in patients taking anticoagulants after valvular surgery: Evaluation by a laser light-scattering method. J. Artif. Organs 2002, 5, 0188–0192. [Google Scholar] [CrossRef]
- Teng, C.M.; Li, H.L.; Wu, T.S.; Huang, S.C.; Huang, T.F. Antiplatelet actions of some coumarin compounds isolated from plant sources. Thromb. Res. 1992, 66, 549–557. [Google Scholar] [CrossRef]
- Gao, C.; Boylan, B.; Fang, J.; Wilcox, D.A.; Newman, D.K.; Newman, P.J. Heparin promotes platelet responsiveness by potentiating αIIbβ3-mediated outside-in signaling. Blood 2011, 117, 4946–4952. [Google Scholar] [CrossRef] [Green Version]
- Chong, B.H.; Ismail, F. The mechanism of heparin-induced platelet aggregation. Eur. J. Haematol. 1989, 43, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Scridon, A.; Şerban, R.C. Laboratory Monitoring: A Turning Point in the Use of New Oral Anticoagulants. Ther. Drug Monit. 2016, 38, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Delinière, A.; Baranchuk, A.; Giai, J.; Bessiere, F.; Maucort-Boulch, D.; Defaye, P.; Marijon, E.; Le Vavasseur, O.; Dobreanu, D.; Scridon, A.; et al. Prediction of ventricular arrhythmias in patients with a spontaneous Brugada type 1 pattern: The key is in the electrocardiogram. Europace 2019, 21, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tarlac, V.; Hamilton, J.R. Using PAR4 Inhibition as an Anti-Thrombotic Approach: Why, How, and When? Int. J. Mol. Sci. 2019, 20, 5629. [Google Scholar] [CrossRef] [Green Version]
- Scridon, A.; Şerban, R.C.; Chevalier, P. Atrial fibrillation: Neurogenic or myogenic? Arch. Cardiovasc. Dis. 2018, 111, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.J.; Reichman, M.E.; Wernecke, M.; Zhang, R.; Southworth, M.R.; Levenson, M.; Sheu, T.C.; Mott, K.; Goulding, M.R.; Houstoun, M.; et al. Cardiovascular, bleeding, and mortality risks in elderly Medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation 2015, 131, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Scridon, A.; Tabib, A.; Barrès, C.; Julien, C.; Chevalier, P. Left atrial endocardial fibrosis and intra-atrial thrombosis—Landmarks of left atrial remodeling in rats with spontaneous atrial tachyarrhythmias. Rom. J. Morphol. Embryol. 2013, 54, 405–411. [Google Scholar] [PubMed]
- Scridon, A.; Perian, M.; Mărginean, A.; Huţanu, A.; Gherţescu, D.; Vântu, A.; Fişcă, P.C.; Chevalier, P.; Şerban, R.C.; Dobreanu, D. Plasma lipids affect dabigatran etexilate anticoagulation in rats with unbalanced diabetes mellitus. J. Diabetes 2018, 10, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Hellkamp, A.S.; Lokhnygina, Y.; Piccini, J.P.; Zhang, Z.; Mohanty, S.; Singer, D.E.; Hacke, W.; Breithardt, G.; Halperin, J.L.; et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: Analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation). J. Am. Coll. Cardiol. 2013, 61, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granger, C.B.; Lopes, R.D.; Hanna, M.; Ansell, J.; Hylek, E.M.; Alexander, J.H.; Thomas, L.; Wang, J.; Bahit, M.C.; Verheugt, F.; et al. Clinical events after transitioning from apixaban versus warfarin to warfarin at the end of the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial. Am. Heart J. 2015, 169, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; von Beckerath, N.; Morath, T.; Stegherr, J.; Mehilli, J.; Sarafoff, N.; Braun, S.; Schulz, S.; Schömig, A.; Kastrati, A. Oral anticoagulation with coumarin derivatives and antiplatelet effects of clopidogrel. Eur. Heart J. 2010, 31, 1205–1211. [Google Scholar] [CrossRef]
- Martischnig, A.M.; Mehilli, J.; Pollak, J.; Petzold, T.; Fiedler, A.K.; Mayer, K.; Schulz-Schüpke, S.; Sibbing, D.; Massberg, S.; Kastrati, A.; et al. Impact of Dabigatran versus Phenprocoumon on ADP Induced Platelet Aggregation in Patients with Atrial Fibrillation with or without Concomitant Clopidogrel Therapy (the Dabi-ADP-1 and Dabi-ADP-2 Trials). Biomed. Res. Int. 2015, 2015, 798486. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alpha-Granules |
| Growth and angiogenic factors (e.g., platelet-derived growth factor, fibroblast growth factor, vascular endothelial growth factor, connective tissue growth factor, epidermal growth factor, transforming growth factor β, insulin-like growth factor 1) |
| Cytokines and chemokines (e.g., interleukin-1β, CD40 ligand, CCL2, CCL3, CCL5, CXCL1, CXCL4, CXCL12, CXCL16, platelet factor 4) |
| Adhesion molecules (e.g., P-selectin, fibronectin, vitronectin, von Willebrand factor) |
| Coagulation factors (e.g., factors V, XIII, von Willebrand factor, high-molecular-weight kininogen, fibrinogen) |
| Anticoagulation factors (e.g., tissue factor pathway inhibitor, protein S) |
| Fibrinolytic factors (e.g., plasmin, plasminogen) |
| Antifibrinolytic factors (e.g., α2-antiplasmin, thrombin activatable fibrinolysis inhibitor, plasminogen activator inhibitor-1) |
| Other molecules (e.g., albumin, calcitonin, angiotensinogen, thrombospondin) |
| Dense granules |
| Serotonin Histamine Adenine nucleotides (ADP, ATP) Cations (e.g., calcium, magnesium), polyphosphate Adhesion molecules (e.g., P-selectin) |
| Lysosomal granules |
| Cathepsin D, E Carboxypeptidase A, B Acid phosphatase Arylsulfatase |
| Cytosol |
| Adhesion molecules (e.g., P-selectin, fibronectin, vitronectin, fibrinogen, thrombospondin, von Willebrand factor) |
| Coagulation factors (e.g., factors V, von Willebrand factor) |
| Platelet activators (e.g., platelet-activating factor, TxA2) |
| Vasoconstrictors (e.g., TxA2, 12-hydroxyeicosatetraenoic acid) |
| Function in Hemostasis | Receptors | Receptor Family | Main Ligands |
|---|---|---|---|
| Platelet adhesion to the vessel wall | GPIb-IX-V * | Leucine-rich repeat | vWF, thrombospondin-1, thrombin, factors XI, XII, P-selectin |
| GPVI | Immunoreceptors | Collagen, laminin | |
| α2β1 | Integrins | Collagen, laminin | |
| α6β1 | Laminin | ||
| α5β1 | Fibronectin | ||
| αVβ3 | Vitronectin, vWF, fibronectin, fibrinogen | ||
| αIIbβ3 (i.e., GPIIb/IIIa) ** | Fibrinogen | ||
| Platelet activation | PAR-1, PAR-4 | G protein-coupled receptors | Thrombin |
| P2Y1, P2Y12 | ADP | ||
| TPα, TPβ | TxA2 | ||
| PGE2 receptor | PGE2 | ||
| 5-HT2A | Serotonin | ||
| P2 × 1 | Ion channel | ATP | |
| Platelet aggregation | Activated αIIbβ3 (i.e., GPIIb/IIIa) | Integrins | Fibrin, vWF, thrombospondin-1, fibronectin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scridon, A. Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 12772. https://doi.org/10.3390/ijms232112772
Scridon A. Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications. International Journal of Molecular Sciences. 2022; 23(21):12772. https://doi.org/10.3390/ijms232112772
Chicago/Turabian StyleScridon, Alina. 2022. "Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications" International Journal of Molecular Sciences 23, no. 21: 12772. https://doi.org/10.3390/ijms232112772
APA StyleScridon, A. (2022). Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications. International Journal of Molecular Sciences, 23(21), 12772. https://doi.org/10.3390/ijms232112772