
The Impact of Sphingosine Kinases on Inflammation-Induced Cytokine Release and Vascular Endothelial Barrier Integrity



Abstract
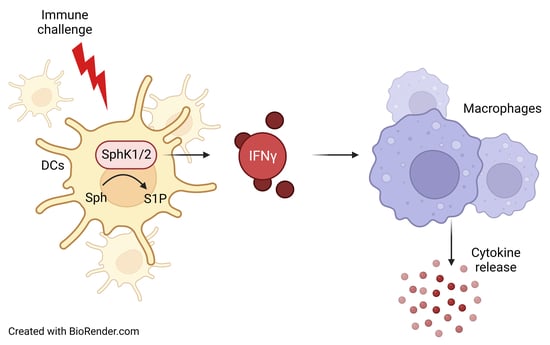
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Altered S1P Levels in SphK1/2 Deficient Mice during Sepsis
2.2. Improved Survival of SphK1/2 Deficient Mice after Systemic Infection
2.3. Similar Impact of Sepsis-Induced Vascular Leakage in SphK1/2 Deficient Mice
2.4. Delayed Onset and Faster Resolution of Pro-Inflammatory Cytokine Release in SphK1/2 Deficient Mice
2.5. Unaltered Cytokine Release in SphK1/2 Deficient Macrophages, Lymphocytes and Lung Endothelial Cells
2.6. Reduced IFN-γ Production by LPS-Stimulated DC from SphK1/2 Deficient Mice Dampened Subsequent Cytokine Release by Macrophages
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Experimental Sepsis
4.3. Plasma Isolation
4.4. Isolation of Bone Marrow Cells from Mice
4.5. Isolation of Splenocytes
4.6. Separation of B and T Cells
4.7. Isolation of Murine Lung Endothelial Cells (MLEC)
4.8. Preparation of L929 Conditioned Medium
4.9. Bone Marrow-Derived Macrophage (BMDM) Differentiation
4.10. Bone Marrow-Derived Dendritic Cell (BMDC) Differentiation
4.11. BMDM Stimulation
4.12. BMDC Stimulation
4.13. T Cell Stimulation
4.14. B Cell Stimulation
4.15. MLEC Stimulation
4.16. Enzyme-Linked Immunosorbent Assay (ELISA)
4.17. Cytometric Bead Array (CBA, BD Biosciences)
4.18. Evans Blue Measurement of Vascular Barrier Integrity
4.19. Lipid Extraction
4.20. Liquid Chromatography Coupled to Triple-Quadrupole Mass Spectrometry (LC-MS/MS)
4.21. Flow Cytometry (FACS)
4.22. Determination of the Bacterial Load
4.23. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Sugiura, M.; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar] [CrossRef] [Green Version]
- Olivera, A.; Kohama, T.; Tu, Z.; Milstien, S.; Spiegel, S. Purification and characterization of rat kidney sphingosine kinase. J. Biol. Chem. 1998, 273, 12576–12583. [Google Scholar] [CrossRef] [Green Version]
- Allende, M.L.; Sasaki, T.; Kawai, H.; Olivera, A.; Mi, Y.; van Echten-Deckert, G.; Hajdu, R.; Rosenbach, M.; Keohane, C.A.; Mandala, S.; et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 2004, 279, 52487–52492. [Google Scholar] [CrossRef] [Green Version]
- Zemann, B.; Kinzel, B.; Muller, M.; Reuschel, R.; Mechtcheriakova, D.; Urtz, N.; Bornancin, F.; Baumruker, T.; Billich, A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 2006, 107, 1454–1458. [Google Scholar] [CrossRef]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef] [Green Version]
- Sensken, S.C.; Bode, C.; Nagarajan, M.; Peest, U.; Pabst, O.; Graler, M.H. Redistribution of sphingosine 1-phosphate by sphingosine kinase 2 contributes to lymphopenia. J. Immunol. 2010, 184, 4133–4142. [Google Scholar] [CrossRef] [Green Version]
- Kharel, Y.; Huang, T.; Salamon, A.; Harris, T.E.; Santos, W.L.; Lynch, K.R. Mechanism of sphingosine 1-phosphate clearance from blood. Biochem. J. 2020, 477, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.R.; Becker, K.P.; Facchinetti, M.M.; Hannun, Y.A.; Obeid, L.M. PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA). J. Biol. Chem. 2002, 277, 35257–35262. [Google Scholar] [CrossRef] [Green Version]
- Blankenbach, K.V.; Claas, R.F.; Aster, N.J.; Spohner, A.K.; Trautmann, S.; Ferreiros, N.; Black, J.L.; Tesmer, J.J.G.; Offermanns, S.; Wieland, T.; et al. Dissecting Gq/11-Mediated Plasma Membrane Translocation of Sphingosine Kinase-1. Cells 2020, 9, 2201. [Google Scholar] [CrossRef]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lepine, S.; Milstien, S.; Spiegel, S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, M.; Kihara, A.; Igarashi, Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 2004, 325, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Camerer, E.; Regard, J.B.; Cornelissen, I.; Srinivasan, Y.; Duong, D.N.; Palmer, D.; Pham, T.H.; Wong, J.S.; Pappu, R.; Coughlin, S.R. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Investig. 2009, 119, 1871–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef]
- Lo, C.G.; Xu, Y.; Proia, R.L.; Cyster, J.G. Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J. Exp. Med. 2005, 201, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Vettorazzi, S.; Bode, C.; Dejager, L.; Frappart, L.; Shelest, E.; Klassen, C.; Tasdogan, A.; Reichardt, H.M.; Libert, C.; Schneider, M.; et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat. Commun. 2015, 6, 7796. [Google Scholar] [CrossRef] [Green Version]
- Niessen, F.; Schaffner, F.; Furlan-Freguia, C.; Pawlinski, R.; Bhattacharjee, G.; Chun, J.; Derian, C.K.; Andrade-Gordon, P.; Rosen, H.; Ruf, W. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 2008, 452, 654–658. [Google Scholar] [CrossRef] [Green Version]
- Avni, D.; Harikumar, K.B.; Sanyal, A.J.; Spiegel, S. Deletion or inhibition of SphK1 mitigates fulminant hepatic failure by suppressing TNFalpha-dependent inflammation and apoptosis. FASEB J. 2021, 35, e21415. [Google Scholar] [CrossRef]
- Mohammed, S.; Vineetha, N.S.; James, S.; Aparna, J.S.; Babu Lankadasari, M.; Maeda, T.; Ghosh, A.; Saha, S.; Li, Q.Z.; Spiegel, S.; et al. Regulatory role of SphK1 in TLR7/9-dependent type I interferon response and autoimmunity. FASEB J. 2020, 34, 4329–4347. [Google Scholar] [CrossRef]
- Weigert, A.; von Knethen, A.; Thomas, D.; Faria, I.; Namgaladze, D.; Zezina, E.; Fuhrmann, D.; Petcherski, A.; Heringdorf, D.M.Z.; Radeke, H.H.; et al. Sphingosine kinase 2 is a negative regulator of inflammatory macrophage activation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.C.; Muller, T.; Graler, M.H. Sphingosine 1-phosphate in sepsis and beyond: Its role in disease tolerance and host defense and the impact of carrier molecules. Cell. Signal. 2021, 78, 109849. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Z.; Feng, X.R.; Borschel, R.H.; Nikolich, M.P.; Feng, J.; Li, Y.S.; Hoover, D.L. HSP-70 mitigates LPS/SKI-induced cell damage by increasing sphingosine kinase 1 (SK1). Prostaglandins Other Lipid Mediat. 2010, 92, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Yao, D.; Zheng, L.; Zhou, Z.; Duan, Y.; Liu, B.; Wang, P.; Li, Y. Sphingosine kinase 1 regulates HMGB1 translocation by directly interacting with calcium/calmodulin protein kinase II-delta in sepsis-associated liver injury. Cell Death Dis. 2020, 11, 1037. [Google Scholar] [CrossRef]
- Zhang, T.; Yan, T.; Du, J.; Wang, S.; Yang, H. Apigenin attenuates heart injury in lipopolysaccharide-induced endotoxemic model by suppressing sphingosine kinase 1/sphingosine 1-phosphate signaling pathway. Chem. Biol. Interact. 2015, 233, 46–55. [Google Scholar] [CrossRef]
- Ugwu, F.N.; Ho, J. Preclinical evidence of sphingosine kinase 1 inhibition in alleviation of intestinal epithelial injury in polymicrobial sepsis. Inflamm. Res. 2019, 68, 723–726. [Google Scholar] [CrossRef]
- Zhong, M.; Wu, W.; Wang, Y.; Mao, H.; Song, J.; Chen, S.; Zhu, D. Inhibition of Sphingosine Kinase 1 Attenuates Sepsis-induced Microvascular Leakage via Inhibiting Macrophage NLRP3 Inflammasome Activation in Mice. Anesthesiology 2020, 132, 1503–1515. [Google Scholar] [CrossRef]
- Liu, Q.; Rehman, H.; Shi, Y.; Krishnasamy, Y.; Lemasters, J.J.; Smith, C.D.; Zhong, Z. Inhibition of sphingosine kinase-2 suppresses inflammation and attenuates graft injury after liver transplantation in rats. PLoS ONE 2012, 7, e41834. [Google Scholar] [CrossRef] [Green Version]
- Joshi, J.C.; Joshi, B.; Rochford, I.; Rayees, S.; Akhter, M.Z.; Baweja, S.; Chava, K.R.; Tauseef, M.; Abdelkarim, H.; Natarajan, V.; et al. SPHK2-Generated S1P in CD11b(+) Macrophages Blocks STING to Suppress the Inflammatory Function of Alveolar Macrophages. Cell Rep. 2020, 30, 4096–4109.e5. [Google Scholar] [CrossRef]
- Fukao, T.; Matsuda, S.; Koyasu, S. Synergistic effects of IL-4 and IL-18 on IL-12-dependent IFN-gamma production by dendritic cells. J. Immunol. 2000, 164, 64–71. [Google Scholar] [CrossRef]
- Stober, D.; Schirmbeck, R.; Reimann, J. IL-12/IL-18-dependent IFN-gamma release by murine dendritic cells. J. Immunol. 2001, 167, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, J.K.; Cornwall, S.M.J.; Musk, A.W.; Alvarez, J.; Mamotte, C.D.S.; Jackaman, C.; Nowak, A.K.; Nelson, D.J. Elderly dendritic cells respond to LPS/IFN-gamma and CD40L stimulation despite incomplete maturation. PLoS ONE 2018, 13, e0195313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, S.; Song, S.B.; Jung, M.; Park, Y.; Bang, J.W.; Kim, T.S.; Park, H.; Kim, C.H.; Yang, Y.H.; Bang, S.I.; et al. Sphingosine kinase inhibitor suppresses IL-18-induced interferon-gamma production through inhibition of p38 MAPK activation in human NK cells. Biochem. Biophys. Res. Commun. 2008, 374, 74–78. [Google Scholar] [CrossRef]
- Jung, I.D.; Lee, J.S.; Kim, Y.J.; Jeong, Y.I.; Lee, C.M.; Baumruker, T.; Billlich, A.; Banno, Y.; Lee, M.G.; Ahn, S.C.; et al. Sphingosine kinase inhibitor suppresses a Th1 polarization via the inhibition of immunostimulatory activity in murine bone marrow-derived dendritic cells. Int. Immunol. 2007, 19, 411–426. [Google Scholar] [CrossRef]
- Furuya, H.; Tamashiro, P.M.; Shimizu, Y.; Iino, K.; Peres, R.; Chen, R.; Sun, Y.; Hannun, Y.A.; Obeid, L.M.; Kawamori, T. Sphingosine Kinase 1 expression in peritoneal macrophages is required for colon carcinogenesis. Carcinogenesis 2017, 38, 1218–1227. [Google Scholar] [CrossRef]
- Ghosh, M.; Thangada, S.; Dasgupta, O.; Khanna, K.M.; Yamase, H.T.; Kashgarian, M.; Hla, T.; Shapiro, L.H.; Ferrer, F.A. Cell-intrinsic sphingosine kinase 2 promotes macrophage polarization and renal inflammation in response to unilateral ureteral obstruction. PLoS ONE 2018, 13, e0194053. [Google Scholar] [CrossRef]
- Yang, G.; Gu, M.; Chen, W.; Liu, W.; Xiao, Y.; Wang, H.; Lai, W.; Xian, G.; Zhang, Z.; Li, Z.; et al. SPHK-2 Promotes the Particle-Induced Inflammation of RAW264.7 by Maintaining Consistent Expression of TNF-alpha and IL-6. Inflammation 2018, 41, 1498–1507. [Google Scholar] [CrossRef]
- Prakash, H.; Luth, A.; Grinkina, N.; Holzer, D.; Wadgaonkar, R.; Gonzalez, A.P.; Anes, E.; Kleuser, B. Sphingosine kinase-1 (SphK-1) regulates Mycobacterium smegmatis infection in macrophages. PLoS ONE 2010, 5, e10657. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Lee, H.J.; Mariko, B.; Lu, Y.C.; Dannenberg, A.J.; Haka, A.S.; Maxfield, F.R.; Camerer, E.; Proia, R.L.; Hla, T. Sphingosine kinases are not required for inflammatory responses in macrophages. J. Biol. Chem. 2013, 288, 32563–32573. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Lu, Z.; Li, Y.; Ru, J.H.; Lopes-Virella, M.F.; Huang, Y. LPS and palmitate synergistically stimulate sphingosine kinase 1 and increase sphingosine 1 phosphate in RAW264.7 macrophages. J. Leukoc. Biol. 2018, 104, 843–853. [Google Scholar] [CrossRef]
- Gan, P.R.; Wang, R.H.; Deng, R.; Wu, H.; Bu, Y.H.; Chen, F.Y.; Dong, X.T.; Ke, J.T. Geniposide inhibits SphK1 membrane targeting to restore macrophage polarization balance in collagen-induced arthritis mice. Eur. J. Pharmacol. 2022, 933, 175271. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.F.; Murray, H.W.; Wiebe, M.E.; Rubin, B.Y. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 1983, 158, 670–689. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauermeister, K.; Burger, M.; Almanasreh, N.; Knopf, H.P.; Schumann, R.R.; Schollmeyer, P.; Dobos, G.J. Distinct regulation of IL-8 and MCP-1 by LPS and interferon-gamma-treated human peritoneal macrophages. Nephrol. Dial. Transplant. 1998, 13, 1412–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef] [PubMed]
- Ramnath, R.D.; Ng, S.W.; Guglielmotti, A.; Bhatia, M. Role of MCP-1 in endotoxemia and sepsis. Int. Immunopharmacol. 2008, 8, 810–818. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Jeon, R.; Vuckovic, I.; Jiang, X.; Lerman, A.; Folmes, C.D.; Dzeja, P.D.; Herrmann, J. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. eBioMedicine 2018, 30, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Romero, C.R.; Herzig, D.S.; Etogo, A.; Nunez, J.; Mahmoudizad, R.; Fang, G.; Murphey, E.D.; Toliver-Kinsky, T.; Sherwood, E.R. The role of interferon-gamma in the pathogenesis of acute intra-abdominal sepsis. J. Leukoc. Biol. 2010, 88, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Gonnert, F.A.; Recknagel, P.; Seidel, M.; Jbeily, N.; Dahlke, K.; Bockmeyer, C.L.; Winning, J.; Losche, W.; Claus, R.A.; Bauer, M. Characteristics of clinical sepsis reflected in a reliable and reproducible rodent sepsis model. J. Surg. Res. 2011, 170, e123–e134. [Google Scholar] [CrossRef]
- Hemdan, N.Y.; Weigel, C.; Reimann, C.M.; Graler, M.H. Modulating sphingosine 1-phosphate signaling with DOP or FTY720 alleviates vascular and immune defects in mouse sepsis. Eur. J. Immunol. 2016, 46, 2767–2777. [Google Scholar] [CrossRef]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression—A new method based on robust nonlinear regression and the false discovery rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thuy, A.V.; Reimann, C.-M.; Ziegler, A.C.; Gräler, M.H. The Impact of Sphingosine Kinases on Inflammation-Induced Cytokine Release and Vascular Endothelial Barrier Integrity. Int. J. Mol. Sci. 2022, 23, 12848. https://doi.org/10.3390/ijms232112848
Thuy AV, Reimann C-M, Ziegler AC, Gräler MH. The Impact of Sphingosine Kinases on Inflammation-Induced Cytokine Release and Vascular Endothelial Barrier Integrity. International Journal of Molecular Sciences. 2022; 23(21):12848. https://doi.org/10.3390/ijms232112848
Chicago/Turabian StyleThuy, Andreas V., Christina-Maria Reimann, Anke C. Ziegler, and Markus H. Gräler. 2022. "The Impact of Sphingosine Kinases on Inflammation-Induced Cytokine Release and Vascular Endothelial Barrier Integrity" International Journal of Molecular Sciences 23, no. 21: 12848. https://doi.org/10.3390/ijms232112848
APA StyleThuy, A. V., Reimann, C. -M., Ziegler, A. C., & Gräler, M. H. (2022). The Impact of Sphingosine Kinases on Inflammation-Induced Cytokine Release and Vascular Endothelial Barrier Integrity. International Journal of Molecular Sciences, 23(21), 12848. https://doi.org/10.3390/ijms232112848