
A Timeframe for SARS-CoV-2 Genomes: A Proof of Concept for Postmortem Interval Estimations


 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Notes on the Reference SARS-CoV-2 Database
2.2. Estimating the Timeframe of a Queried Genome

{kind=link}
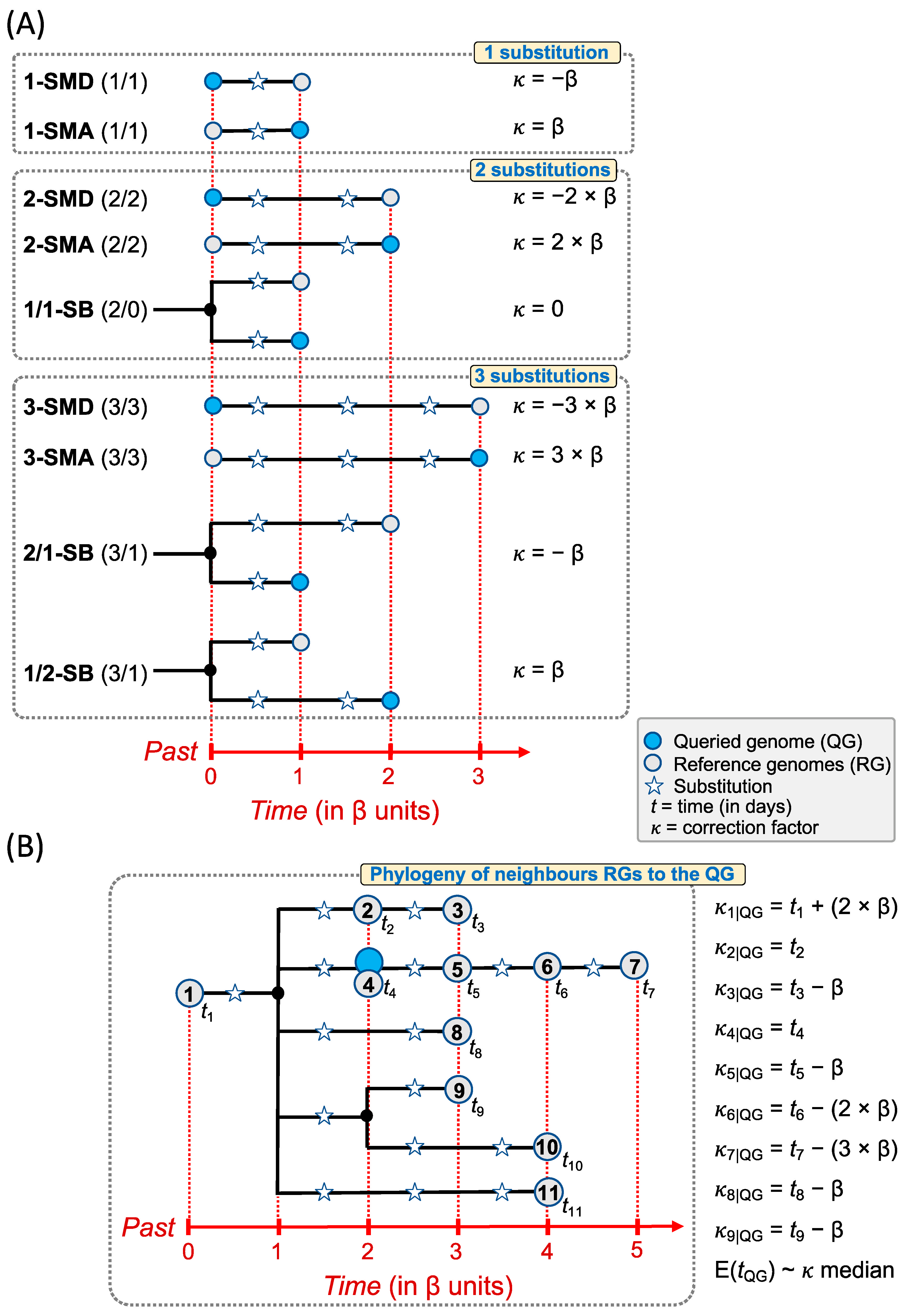{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models * | Correction | Median | 1st Quartile | 2nd Quartile | Missing | Outliers |
|---|---|---|---|---|---|---|
| Simple models | ||||||
| RG = QG | NC | 0 | −8 | 5.5 | 74.4 | 10.5 |
| 1-SMD (1/1) | NC | 6 | −3 | 19 | 79.7 | 5.8 |
| b = 7 | −1 | −10 | 12 | 79.7 | 5.8 | |
| b = 16 | −10 | −19 | 3 | 79.7 | 5.8 | |
| 1-SMA (1/1) | NC | −4 | −17.5 | 3 | 71.5 | 5.8 |
| b = 7 | −11 | −24.5 | −4 | 71.5 | 5.8 | |
| b = 16 | −20 | −33.5 | −13 | 71.5 | 5.8 | |
| 2-SMD (2/2) | NC | 10 | −2 | 25 | 83.3 | 4.6 |
| b = 7 | −4 | −16 | 11 | 83.3 | 4.6 | |
| b = 16 | −22 | −34 | −7 | 83.3 | 4.6 | |
| 2-SMA (2/2) | NC | −6 | −22 | 1 | 69.3 | 5.2 |
| b = 7 | 8 | −8 | 15 | 69.3 | 5.2 | |
| b = 16 | 26 | 10 | 33 | 69.3 | 5.2 | |
| 1/1-SB (2/0) | NC | 2 | −10 | 14 | 75.5 | 5.1 |
| 3-SMD (3/3) | NC | 16 | 1 | 32 | 87.3 | 5.2 |
| b = 7 | −5 | −20 | 11 | 87.3 | 5.2 | |
| b = 16 | −32 | −47 | −16 | 87.3 | 5.2 | |
| 3-SMA (3/3) | NC | −10.5 | −28 | 0 | 73.0 | 4.5 |
| b = 7 | 10.5 | −7 | 21 | 73.0 | 4.5 | |
| b = 16 | 37.5 | 20 | 48 | 73.0 | 4.5 | |
| 2/1-SB (3/1) | NC | −1 | −16 | 10 | 74.1 | 4.1 |
| b = 7 | 6 | −9 | 17 | 74.1 | 4.1 | |
| b = 16 | 15 | 0 | 26 | 74.1 | 4.1 | |
| 1/2-SB (3/1) | NC | 6 | −8 | 21 | 79.6 | 4.7 |
| b = 7 | −1 | −15 | 14 | 79.6 | 4.7 | |
| b = 16 | −10 | −24 | 5 | 79.6 | 4.7 | |
| Mixed models | ||||||
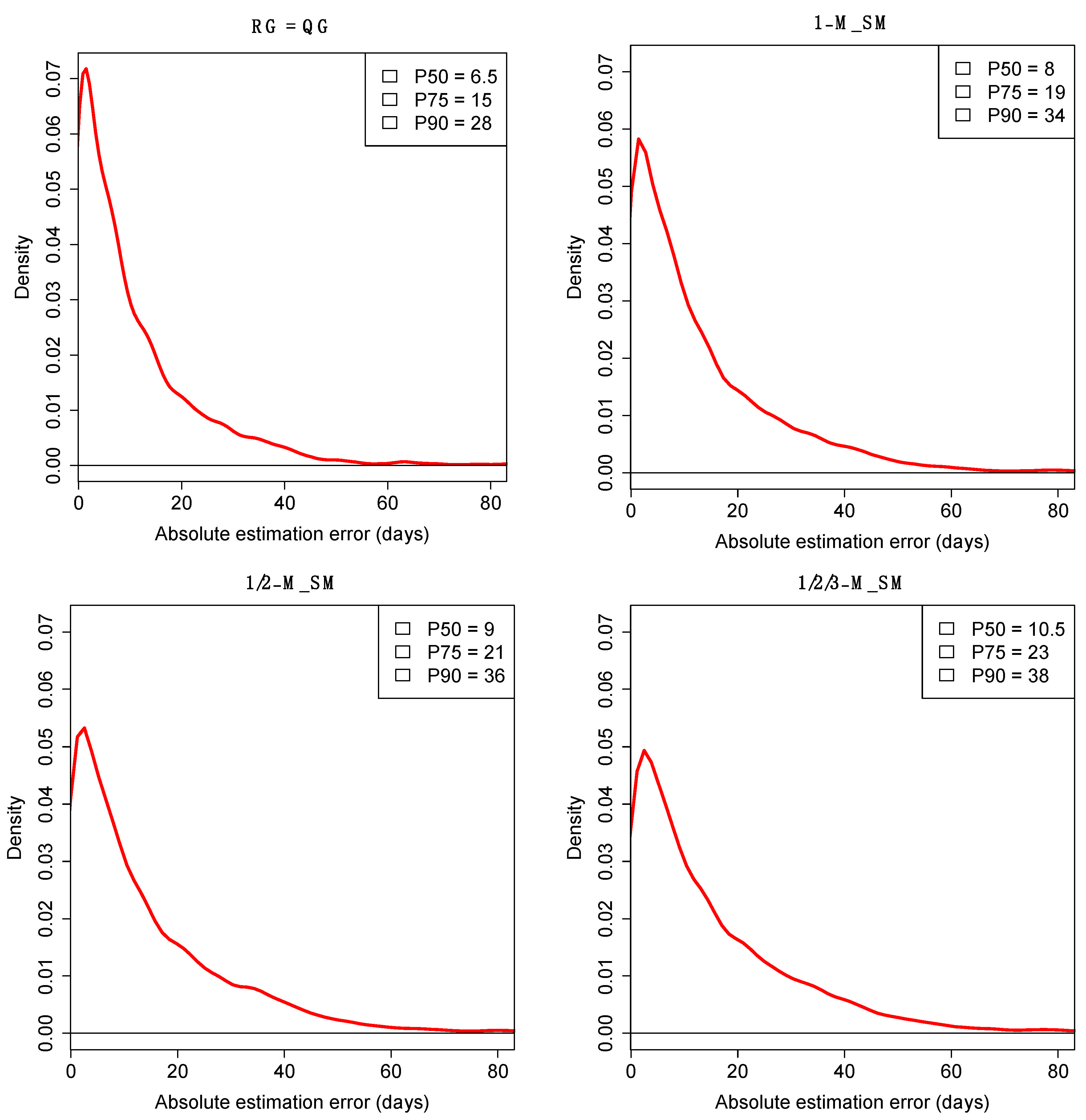
| 1-M_SM | NC | 0 | −8 | 5.5 | 74.4 | 10.5 |
| b = 7 | −0.5 | −12.5 | 5.5 | 53.2 | 7.5 | |
| b = 16 | 0 | −11 | 7 | 53.2 | 7.4 | |
| 1/2-M_SM | NC | 1 | −9 | 9 | 53.2 | 7.2 |
| b = 7 | −1.5 | −15 | 5 | 31.4 | 6 | |
| b = 16 | 2 | −9 | 13 | 31.4 | 4.6 | |
| 1/2/3-M_SM | NC | 7 | −5 | 24 | 31.4 | 2.4 |
| b = 7 | −3 | −17 | 5 | 16.5 | 5.6 | |
| b = 16 | 4 | −9 | 15 | 16.5 | 4 | |
2.3. Error Associated to the tE-QG Estimates
2.4. The Effect of Database Geographic Coverage
3. Discussion
- (i)
- The RNA of a microorganisms of interest might not be present, or be too degraded, in the corpse or the biological sample of forensic interest.
- (ii)
- A large reference genome database is always mandatory for the microorganism of interest.
- (iii)
- The method is probably sensitive to missing or incorrect data (in both the reference database and the interrogated genomes); however, the simulations carried out by Turaknhia et al. [23] in UShER suggested that time estimates could experience only minor deviations.
- (i)
- It has potential for the estimation of time intervals running from about one month in the past to a (very) distant past (of years ago), which is the time range not covered by current methods (e.g., present-day methods for the estimation of the PMI only work for periods of just a few hours/days in the past).
- (ii)
- It is highly precise because even for several years old specimens it may allow to obtain short error estimates (measured as IQRs) of only a few days/weeks; moreover, the IQRs are approximately constant if the reference database keeps a considerable sample size over time (e.g., in the order of the one employed in the present study).
- (iii)
- It would work for symptomatic or asymptomatic clinical cases [36] and it does not depend on the severity and body tissue [37] analyzed because the only information that is needed for the analysis is the genome sequence of the microorganism, independently of the clinical manifestations of the infected individual.
- (iv)
- The procedure performs well in circumstances where the QGs do not have a well-represented local reference genome database. A possible explanation for this may be that the SARS-CoV-2 phylogenetic tree is so large that the deficient contribution of some countries to the database might be phylogenetically compensated by the large contribution of a few countries.
- (v)
- It is cost effective because it would only require sequencing the whole genome of the microorganism using well-established NGS techniques (currently available in, e.g., most hospital settings and research institutions).
- (vi)
- Theoretically, the SARS-CoV-2 VMCD method would work even if more than one strain is present because all the infected strains in an individual should have comparable ‘evolutionary times’ (moreover, note that usually it is the consensus genome sequence of the virus that is reported in an individual and in genome repositories).
- (vii)
- The ultrafast sample placement of QGs in a preexisting phylogeny provides an agile and computationally easy environment for real casework without specialized personnel.
- (viii)
- The method is not sensitive to environmental factors (beyond the effect these factors could have in degrading the genetic material and precluding its sequencing).
- (ix)
- More than one pathogen could be used to obtain independent estimates of infection times.
4. Material and Methods
4.1. The SARS-CoV-2 Genome Database
4.2. Phylogenetic Allocation of Genomes into the SARS-CoV-2 Tree
4.3. Basic Time Estimates for Queried Genomes
4.4. Corrected Time Estimates for Queried Genomes Using the Molecular Clock
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sampaio-Silva, F.; Magalhaes, T.; Carvalho, F.; Dinis-Oliveira, R.J.; Silvestre, R. Profiling of RNA degradation for estimation of post mortem [corrected] interval. PLoS ONE 2013, 8, e56507. [Google Scholar] [CrossRef] [PubMed]
- Scrivano, S.; Sanavio, M.; Tozzo, P.; Caenazzo, L. Analysis of RNA in the estimation of post-mortem interval: A review of current evidence. Int. J. Leg. Med. 2019, 133, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Dell’Aquila, M.; De Matteis, A.; Scatena, A.; Costantino, A.; Camporeale, M.C.; De Filippis, A. Estimation of the time of death: Where we are now? Clin. Ter. 2021, 172, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xing, Y.; Jiang, P.; Gan, L.; Zhao, F.; Peng, W.; Li, W.; Tong, Y.; Deng, S. Predicting the postmortem interval using human intestinal microbiome data and random forest algorithm. Sci. Justice 2021, 61, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.R.; Trinidad, D.D.; Guzman, S.; Khan, Z.; Parziale, J.V.; DeBruyn, J.M.; Lents, N.H. A Machine Learning Approach for Using the Postmortem Skin Microbiome to Estimate the Postmortem Interval. PLoS ONE 2016, 11, e0167370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, J.L. Estimating the postmortem interval using microbes: Knowledge gaps and a path to technology adoption. Forensic Sci. Int. Genet. 2019, 38, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciaffi, R.; Feola, A.; Perfetti, E.; Manciocchi, S.; Potenza, S.; Marella, G.L. Overview on the estimation of post mortem interval in forensic anthropology: Review of the literature and practical experience. Rom. J. Leg. Med. 2018, 26, 403–411. [Google Scholar]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S. Molecular clocks: Four decades of evolution. Nat. Rev. Genet. 2005, 6, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Capodiferro, M.R.; Aram, B.; Raveane, A.; Rambaldi Migliore, N.; Colombo, G.; Ongaro, L.; Rivera, J.; Mendizabal, T.; Hernandez-Mora, I.; Tribaldos, M.; et al. Archaeogenomic distinctiveness of the Isthmo-Colombian area. Cell 2021, 184, 1706–1723.e24. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.; Sikora, M.; Gopalakrishnan, S.; Cassidy, L.M.; Maisano Delser, P.; Sandoval Velasco, M.; Schraiber, J.G.; Rasmussen, S.; Homburger, J.R.; Avila-Arcos, M.C.; et al. Origins and genetic legacies of the Caribbean Taino. Proc. Natl. Acad. Sci. USA 2018, 115, 2341–2346. [Google Scholar] [CrossRef] [PubMed]
- Brandini, S.; Bergamaschi, P.; Cerna, M.F.; Gandini, F.; Bastaroli, F.; Bertolini, E.; Cereda, C.; Ferretti, L.; Gomez-Carballa, A.; Battaglia, V.; et al. The Paleo-Indian Entry into South America According to Mitogenomes. Mol. Biol. Evol. 2018, 35, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Carballa, A.; Pardo-Seco, J.; Brandini, S.; Achilli, A.; Perego, U.A.; Coble, M.D.; Diegoli, T.M.; Álvarez-Iglesias, V.; Martinón-Torres, F.; Olivieri, A.; et al. The peopling of South America and the trans-Andean gene flow of the first settlers. Genome Res. 2018, 28, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Martinón-Torres, F.; Salas, A. Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res. 2020, 30, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Pérez Del Molino, M.L.; Martinón-Torres, F.; Salas, A. Phylogeography of SARS-CoV-2 pandemic in Spain: A story of multiple introductions, micro-geographic stratification, founder effects, and super-spreaders. Zool Res. 2020, 41, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Seco, J.; Gomez-Carballa, A.; Bello, X.; Martinon-Torres, F.; Salas, A. Pitfalls of barcodes in the study of worldwide SARS-CoV-2 variation and phylodynamics. Zool Res. 2021, 42, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. SARS-CoV-2 (COVID-19) by the numbers. Elife 2020, 9, e57309. [Google Scholar] [CrossRef] [Green Version]
- Bello, X.; Pardo-Seco, J.; Gomez-Carballa, A.; Weissensteiner, H.; Martinon-Torres, F.; Salas, A. CovidPhy: A tool for phylogeographic analysis of SARS-CoV-2 variation. Environ. Res. 2022, 204, 111909. [Google Scholar] [CrossRef]
- Gomez-Carballa, A.; Pardo-Seco, J.; Bello, X.; Martinon-Torres, F.; Salas, A. Superspreading in the emergence of COVID-19 variants. Trends Genet. 2021, 37, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Jarvis, C.I.; Group, C.C.-W.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Beltempo, P.; Curti, S.M.; Maserati, R.; Gherardi, M.; Castelli, M. Persistence of SARS-CoV-2 RNA in post-mortem swab 35 days after death: A case report. Forensic Sci. Int. 2021, 319, 110653. [Google Scholar] [CrossRef] [PubMed]
- Sablone, S.; Solarino, B.; Ferorelli, D.; Benevento, M.; Chironna, M.; Loconsole, D.; Sallustio, A.; Dell’Erba, A.; Introna, F. Post-mortem persistence of SARS-CoV-2: A preliminary study. Forensic Sci. Med. Pathol. 2021, 17, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Rosato, E.; Locatelli, M.; Tartaglia, A.; Falco, P.; Petrarca, C.; Potenza, F.; Damiani, V.; Mandatori, D.; De Laurenzi, V.; et al. Long persistence of severe acute respiratory syndrome coronavirus 2 swab positivity in a drowned corpse: A case report. J. Med. Case Rep. 2022, 16, 72. [Google Scholar] [CrossRef]
- Heinrich, F.; Meissner, K.; Langenwalder, F.; Puschel, K.; Norz, D.; Hoffmann, A.; Lutgehetmann, M.; Aepfelbacher, M.; Bibiza-Freiwald, E.; Pfefferle, S.; et al. Postmortem Stability of SARS-CoV-2 in Nasopharyngeal Mucosa. Emerg. Infect. Dis. 2021, 27, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Arena, V.; Cattani, P.; Dell’Aquila, M.; Liotti, F.M.; Sanguinetti, M.; Oliva, A.; Gemelli against COVID-19 Group. SARS-CoV-2 viral load and replication in postmortem examinations. Int. J. Leg. Med. 2022, 136, 935–939. [Google Scholar] [CrossRef]
- White, K.; Yang, P.; Li, L.; Farshori, A.; Medina, A.E.; Zielke, H.R. Effect of Postmortem Interval and Years in Storage on RNA Quality of Tissue at a Repository of the NIH NeuroBioBank. Biopreserv. Biobank 2018, 16, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Smith, O.; Clapham, A.; Rose, P.; Liu, Y.; Wang, J.; Allaby, R.G. A complete ancient RNA genome: Identification, reconstruction and evolutionary history of archaeological Barley Stripe Mosaic Virus. Sci. Rep. 2014, 4, 4003. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Solis, A.A.; Villa-Islas, V.; Bravo-Lopez, M.J.; Sandoval-Velasco, M.; Wesp, J.K.; Gomez-Valdes, J.A.; Moreno-Cabrera, M.L.; Meraz, A.; Solis-Pichardo, G.; Schaaf, P.; et al. Ancient viral genomes reveal introduction of human pathogenic viruses into Mexico during the transatlantic slave trade. Elife 2021, 10, e68612. [Google Scholar] [CrossRef] [PubMed]
- Smith, O.; Dunshea, G.; Sinding, M.S.; Fedorov, S.; Germonpre, M.; Bocherens, H.; Gilbert, M.T.P. Ancient RNA from Late Pleistocene permafrost and historical canids shows tissue-specific transcriptome survival. PLoS Biol. 2019, 17, e3000166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvignac-Spencer, S.; Dux, A.; Gogarten, J.F.; Patrono, L.V. Molecular archeology of human viruses. Adv. Virus Res. 2021, 111, 31–61. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T. Chronumental: Time tree estimation from very large phylogenies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Howell, N.; Smejkal, C.B.; Mackey, D.A.; Chinnery, P.F.; Turnbull, D.M.; Herrnstadt, C. The pedigree rate of sequence divergence in the human mitochondrial genome: There is a difference between phylogenetic and pedigree rates. Am. J. Hum. Genet. 2003, 72, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Nioi, M.; Napoli, P.E.; Fossarello, M.; d’Aloja, E. Autopsies and Asymptomatic Patients During the COVID-19 Pandemic: Balancing Risk and Reward. Front. Public Health 2020, 8, 595405. [Google Scholar] [CrossRef]
- Gómez-Carballa, A.; Rivero-Calle, I.; Pardo-Seco, J.; Gómez-Rial, J.; Rivero-Velasco, C.; Rodriguez-Nunez, N.; Barbeito-Castineiras, G.; Pérez-Freixo, H.; Cebey-López, M.; Barral-Arca, R.; et al. A multi-tissue study of immune gene expression profiling highlights the key role of the nasal epithelium in COVID-19 severity. Environ. Res. 2022, 210, 112890. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.H.; Porter, A.F.; Wirth, W.; Duchene, S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol. Biol Evol. 2022, 39, msac013. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 Variants in Patients with Immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef]
- Weigang, S.; Fuchs, J.; Zimmer, G.; Schnepf, D.; Kern, L.; Beer, J.; Luxenburger, H.; Ankerhold, J.; Falcone, V.; Kemming, J.; et al. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat. Commun. 2021, 12, 6405. [Google Scholar] [CrossRef]
- Prescott, J.; Bushmaker, T.; Fischer, R.; Miazgowicz, K.; Judson, S.; Munster, V.J. Postmortem stability of Ebola virus. Emerg. Infect. Dis. 2015, 21, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Schuenemann, V.J.; Avanzi, C.; Krause-Kyora, B.; Seitz, A.; Herbig, A.; Inskip, S.; Bonazzi, M.; Reiter, E.; Urban, C.; Dangvard Pedersen, D.; et al. Ancient genomes reveal a high diversity of Mycobacterium leprae in medieval Europe. PLoS Pathog. 2018, 14, e1006997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtwangler, A.; Neukamm, J.; Bohme, L.; Reiter, E.; Vollstedt, M.; Arora, N.; Singh, P.; Cole, S.T.; Knauf, S.; Calvignac-Spencer, S.; et al. Comparison of target enrichment strategies for ancient pathogen DNA. Biotechniques 2020, 69, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Gabbrielli, M.; Gandolfo, C.; Anichini, G.; Candelori, T.; Benvenuti, M.; Savellini, G.G.; Cusi, M.G. How long can SARS-CoV-2 persist in human corpses? Int. J. Infect. Dis. 2021, 106, 1–2. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, A.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Sheehan, S.A.; Hamilton, K.L.; Retzbach, E.P.; Balachandran, P.; Krishnan, H.; Leone, P.; Goldberg, G.S. Evidence that Maackia amurensis seed lectin (MASL) exerts pleiotropic actions on oral squamous cells to inhibit SARS-CoV-2 infection and COVID-19 disease progression. Cell Res. 2021, 403, 112594. [Google Scholar] [CrossRef] [PubMed]
- Deinhardt-Emmer, S.; Wittschieber, D.; Sanft, J.; Kleemann, S.; Elschner, S.; Haupt, K.F.; Vau, V.; Haring, C.; Rodel, J.; Henke, A.; et al. Early postmortem mapping of SARS-CoV-2 RNA in patients with COVID-19 and the correlation with tissue damage. Elife 2021, 10, e60361. [Google Scholar] [CrossRef]
- R core Team. R: A Language and Enviroment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardo-Seco, J.; Bello, X.; Gómez-Carballa, A.; Martinón-Torres, F.; Muñoz-Barús, J.I.; Salas, A. A Timeframe for SARS-CoV-2 Genomes: A Proof of Concept for Postmortem Interval Estimations. Int. J. Mol. Sci. 2022, 23, 12899. https://doi.org/10.3390/ijms232112899
Pardo-Seco J, Bello X, Gómez-Carballa A, Martinón-Torres F, Muñoz-Barús JI, Salas A. A Timeframe for SARS-CoV-2 Genomes: A Proof of Concept for Postmortem Interval Estimations. International Journal of Molecular Sciences. 2022; 23(21):12899. https://doi.org/10.3390/ijms232112899
Chicago/Turabian StylePardo-Seco, Jacobo, Xabier Bello, Alberto Gómez-Carballa, Federico Martinón-Torres, José Ignacio Muñoz-Barús, and Antonio Salas. 2022. "A Timeframe for SARS-CoV-2 Genomes: A Proof of Concept for Postmortem Interval Estimations" International Journal of Molecular Sciences 23, no. 21: 12899. https://doi.org/10.3390/ijms232112899
APA StylePardo-Seco, J., Bello, X., Gómez-Carballa, A., Martinón-Torres, F., Muñoz-Barús, J. I., & Salas, A. (2022). A Timeframe for SARS-CoV-2 Genomes: A Proof of Concept for Postmortem Interval Estimations. International Journal of Molecular Sciences, 23(21), 12899. https://doi.org/10.3390/ijms232112899