
Neurodegenerative Diseases: Can Caffeine Be a Powerful Ally to Weaken Neuroinflammation?

 ,
,  ,
, 
Abstract
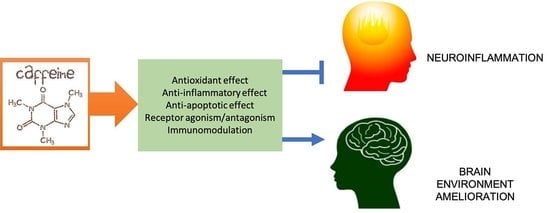
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
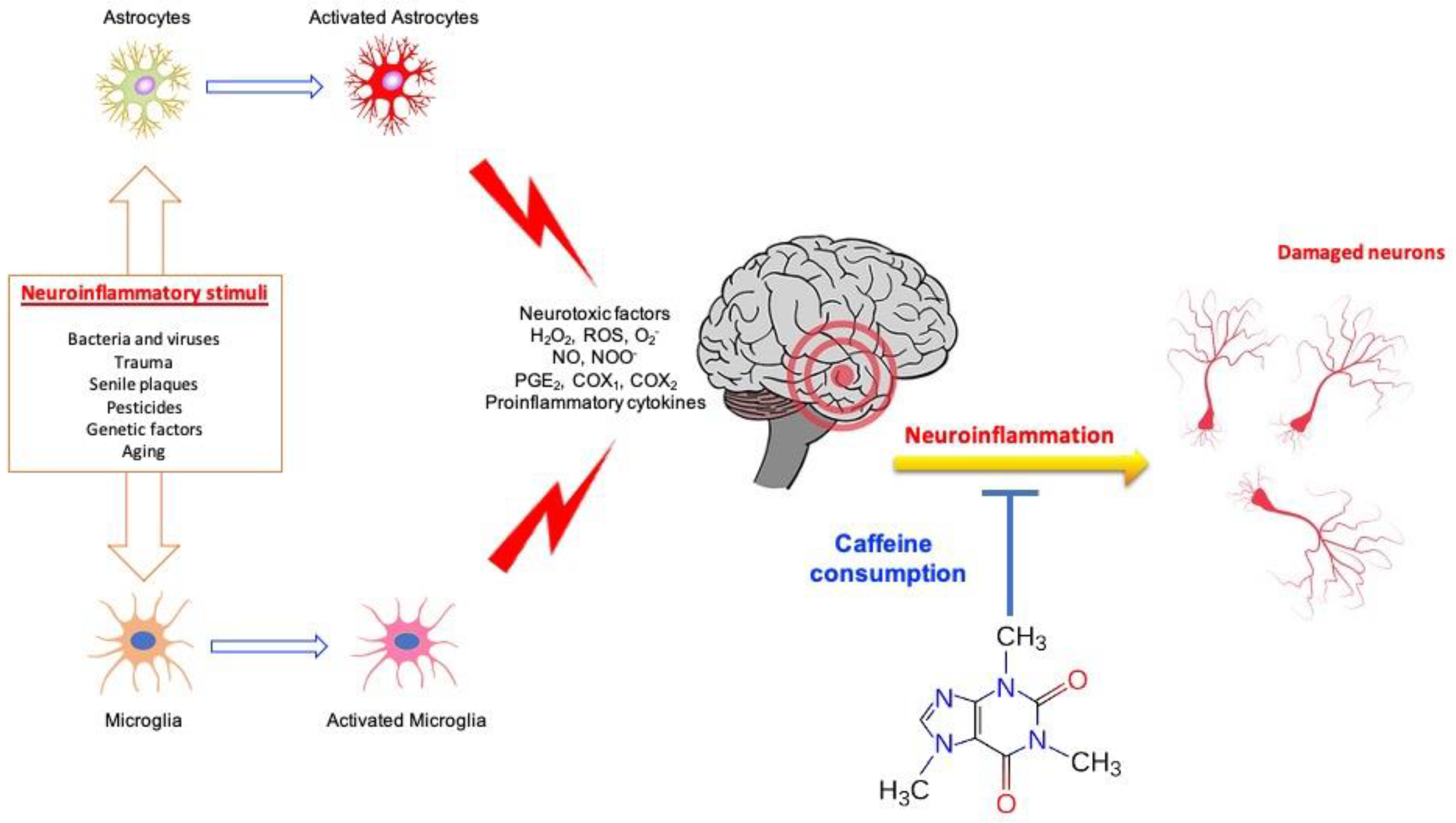
2. Neuroinflammation, Neurodegeneration and Caffeine
3. Caffeine and Glial Cells
4. Neuroprotective Effect of Caffeine in Neuroinflammatory and Neurodegenerative Diseases
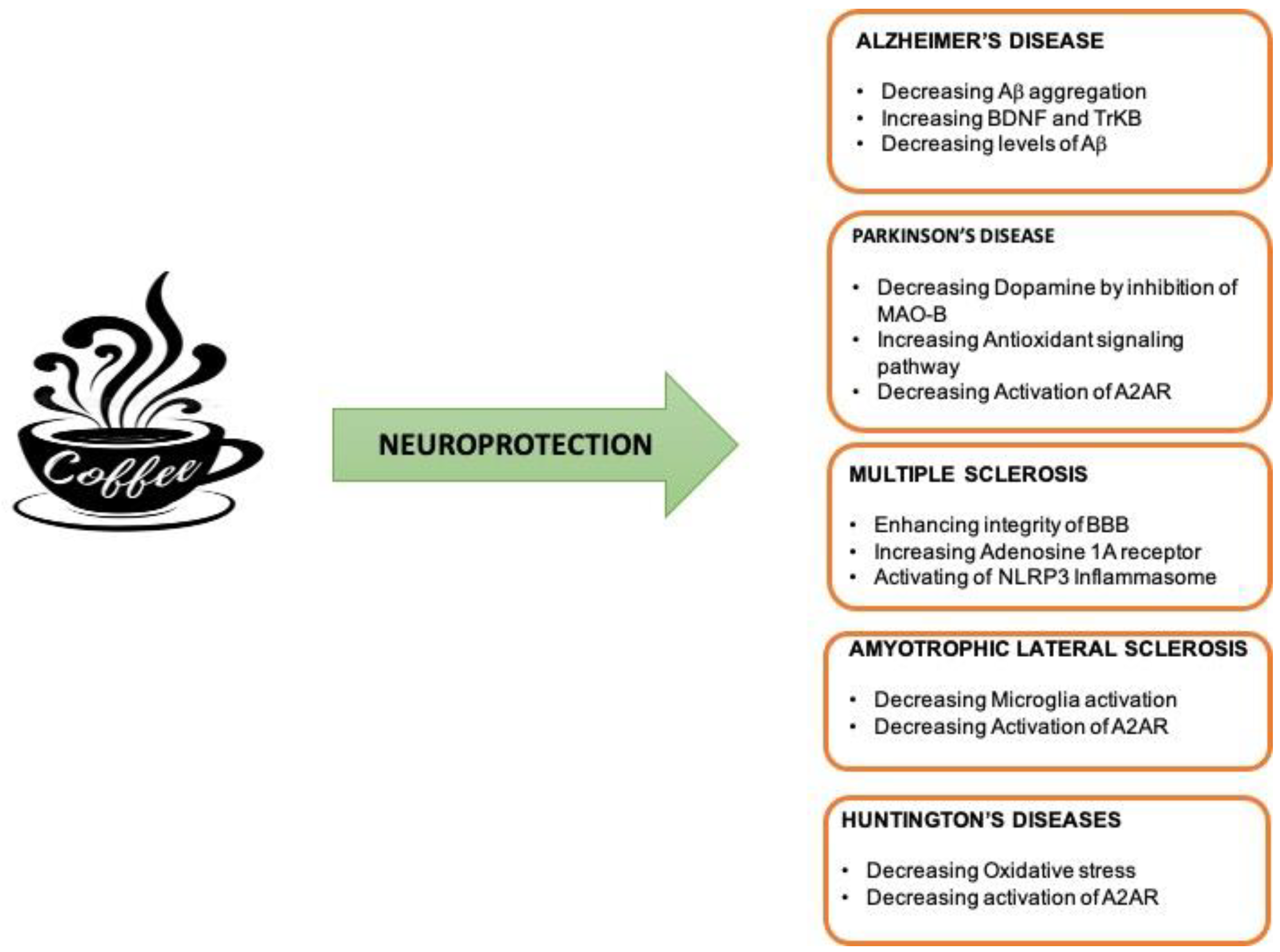
4.1. Neuroprotective Effect of Caffeine in AD
4.2. Neuroprotective Effect of Caffeine in PD
4.3. Neuroprotective Effect of Caffeine in MS
4.4. Neuroprotective Effect of Caffeine in ALS
4.5. Neuroprotective Effect of Caffeine in HD
5. Controlled Release Systems for Caffeine
6. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer’s Res. Ther. 2017, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Inflammatory neuroprotection following traumatic brain injury. Science 2016, 353, 783–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in neuroinflammation and neurodegeneration: From understanding to therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Porro, C.; Calvello, R.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Microglia Mediated Neuroinflammation: Focus on PI3K Modulation. Biomolecules 2020, 10, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, T.; Boddeke, H.W.G.M. Glial cells as drug targets: What does it take? GLIA 2016, 64, 1742–1754. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pranata, R.; Feraldho, A.; Lim, M.A.; Henrina, J.; Vania, R.; Golden, N.; July, J. Coffee and tea consumption and the risk of glioma: A systematic review and dose-response meta-analysis. Br. J. Nutr. 2022, 127, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Szopa, A.; Serefko, A.; Poleszak, E.; Wlaź, P. Neuroprotective Effects of Coffee Bioactive Compounds: A Review. Int. J. Mol. Sci. 2021, 22, 107. [Google Scholar] [CrossRef] [PubMed]
- Rodak, K.; Kokot, I.; Kratz, E.M. Caffeine as a Factor Influencing the Functioning of the Human Body—Friend or Foe? Nutrients 2021, 13, 3088. [Google Scholar] [CrossRef]
- Grzegorzewski, J.; Bartsch, F.; Köller, A.; König, M. Pharmacokinetics of Caffeine: A Systematic Analysis of Reported Data for Application in Metabolic Phenotyping and Liver Function Testing. Front. Pharmacol. 2022, 12, 752826. [Google Scholar] [CrossRef] [PubMed]
- Che, B.; Wang, L.; Zhang, Z.; Zhang, Y.; Deng, Y. Distribution and accumulation of caffeine in rat tissues and its inhibition on semicarbazide-sensitive amine oxidase. Neurotoxicology 2012, 33, 1248–1253. [Google Scholar] [CrossRef]
- Ren, X.; Chen, J.F. Caffeine and Parkinson’s Disease: Multiple Benefits and Emerging Mechanisms. Front. Neurosci. 2020, 14, 602697. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Jiang, D.; Liu, P.; Ge, Y.; Moghekar, A.; Lu, H. Blood-brain barrier permeability in response to caffeine challenge. Magn. Reson. Med. 2022, 88, 2259–2266. [Google Scholar] [CrossRef] [PubMed]
- Graneri, L.; Lam, V.; D’Alonzo, Z.; Nesbit, M.; Mamo, J.C.L.; Takechi, R. The Consumption of Energy Drinks Induces Blood-Brain Barrier Dysfunction in Wild-Type Mice. Front. Nutr. 2021, 3, 668514. [Google Scholar] [CrossRef] [PubMed]
- Reis, R.; Charehsaz, M.; Sipahi, H.; Ekici, A.I.; Macit, Ç.; Akkaya, H.; Aydın, A. Energy Drink Induced Lipid Peroxidation and Oxidative Damage in Rat Liver and Brain When Used Alone or Combined with Alcohol. J. Food Sci. 2017, 82, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Battig, K.; Holmen, J.; Nehlig, A.; Zvartau, E.E. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 1999, 51, 83–133. [Google Scholar] [PubMed]
- Lopes, J.P.; Pliássova, A.; Cunha, R.A. The physiological effects of caffeine on synaptic transmission and plasticity in the mouse hippocampus selectively depend on adenosine A 1 and A 2A receptors. Biochem. Pharmacol. 2019, 166, 313–321. [Google Scholar] [CrossRef]
- Herden, L.; Weissert, R. The Impact of Coffee and Caffeine on Multiple Sclerosis Compared to Other Neurodegenerative Diseases. Front. Nutr. 2018, 5, 133. [Google Scholar] [CrossRef] [Green Version]
- Pourshahidi, K.L.; Navarini, L.; Petracco, M.; Strain, J.J. A Comprehensive Overview of the Risks and Benefits of Coffee Consumption. Compr. Rev. Food Sci. Food Saf. 2016, 15, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Mena, P.; Tassott, M.; Martini, D.; Rosi, A.; Brighenti, F.; Del Rio, D. The Pocket-4-Life project, bioavailability and beneficial properties of the bioactive compounds of espresso coffee and cocoa-based confectionery containing coffee: Study protocol for a randomized cross-over trial. Trials 2017, 18, 527. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, T.G.; Popescu, B.O. Impact of Caffeine on Alzheimer’s Disease Pathogenesis-Protective or Risk Factor? Life 2022, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.; Grotta, J.C.; Aronowski, J. Combination of low dose ethanol and caffeine protects brain from damage produced by focal ischemia in rats. Neuropharmacology 2000, 39, 515–522. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, L. The Neuroprotective Effects of Moderate and Regular Caffeine Consumption in Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2021, 2021, 5568011. [Google Scholar] [CrossRef]
- Hernández, E.R.; Olvera, B.A.; González, D.C.; Marín, O.G.; Mercado, D.P.; Gil, L.V.; Hernández-Zimbrón, L.F.; Salgado, S.J.L.; Limón, I.D.; Zenteno, E. Neuroinflammation and galectins: A key relationship in neurodegenerative diseases. Glycoconj. J. 2022, 39, 685–699. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Calvello, R.; Ruggiero, M.; Panaro, M.A. Inflammaging and Brain: Curcumin and Its Beneficial Potential as Regulator of Microglia Activation. Molecules 2022, 27, 341. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Kell, D.B.; Pretorius, E. The Role of Lipopolysaccharide-Induced Cell Signalling in Chronic Inflammation. Chronic Stress 2022, 6, 24705470221076390. [Google Scholar] [CrossRef] [PubMed]
- Feehan, J.; Tripodi, N.; Apostolopoulos, V. The twilight of the immune system: The impact of immunosenescence in aging. Maturitas 2021, 147, 7–13. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Jo, M.G.; Kim, S.Y.; Chung, G.C.; Lee, S.B. Dietary Antioxidants and the Mitochondrial Quality Control: Their Potential Roles in Parkinson’s Disease Treatment. Antioxidants 2020, 9, E1056. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and Oxidative Stress in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darshpreet, K.; Sharma, V.; Deshmukh, R. Activation of Microglia and Astrocytes: A Roadway to Neuroinflammation and Alzheimer’s Disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Giacomelli, C.; Martini, C. Brain ageing and neurodegenerative disease: The role of cellular waste management. Biochem. Pharmacol. 2018, 158, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolahdouzan, M.; Mazen, J.; Hamadeh, M.J. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and microglia: In sickness and in health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef] [PubMed]
- de Haas, A.H.; Boddeke, H.W.; Biber, K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 2008, 56, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Torre, M.E.; Ines Villano, I.; Monda, M.; Messina, M.; Cibelli, G.; Valenzano, A.; Pisanelli, D.; Panaro, M.A.; Tartaglia, N.; Ambrosi, A.; et al. Role of Vitamin E and the Orexin System in Neuroprotection. Brain Sci. 2021, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Calvello, R.; Porro, C.; Trotta, T.; Panaro, M.A. Understanding the role of SOCS signaling in neurodegenerative diseases: Current and emerging concepts. Cytokine Growth Factor Rev. 2017, 37, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Microglia and astrocytes in Alzheimer’s disease: Implications for therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Madeira, M.H.; Boia, R.; Ambrósio, A.F.; Santiago, A.R. Having a Coffee Break: The Impact of Caffeine Consumption on Microglia-Mediated Inflammation in Neurodegenerative Diseases. Mediat. Inflamm. 2017, 2017, 4761081. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Gupta, S.P.; Srivastava, G.; Srivastava, P.K.; Singh, M.P. Role of secondary mediators in caffeine-mediated neuroprotection in maneb- and paraquat-induced Parkinson’s disease phenotype in the mouse. Neurochem. Res. 2012, 37, 875–884. [Google Scholar] [CrossRef]
- Kang, C.H.; Jayasooriya, R.G.P.T.; Dilshara, M.G.; Choi, Y.H.; Jeong, Y.K.; Kim, N.D.; Kim, G.Y. Caffeine suppresses lipopolysaccharide-stimulated BV2 microglial cells by suppressing Akt-mediated NF-κB activation and ERK phosphorylation. Food Chem. Toxicol. 2012, 50, 4270–4276. [Google Scholar] [CrossRef]
- Badshah, H.; Ikram, M.; Ali, W.; Ahmad, S.; Hahm, J.R.; Kim, M.O. Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains. Biomolecules 2019, 9, 719. [Google Scholar] [CrossRef] [Green Version]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Nobre, H.V., Jr.; de Andrade Cunha, G.M.; de Vasconcelos, L.M.; Magalhães, H.I.F.; Neto, R.N.O.; Maia, F.D.; de Motaes, M.O.; Moreira Leal, L.K.A.; de Barros Viana, G.S. Caffeine and CSC, adenosine A2A antagonists, offer neuroprotection against 6-OHDA-induced neurotoxicity in rat mesencephalic cells. Neurochem. Int. 2010, 56, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.F. Chronic Caffeine Treatment Protects Against α-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Medina, J.; Pinto-Xavier, A.; Rodríguez-Arias, M.; Miñarro, J.; Valverde, O. Influence of chronic caffeine on MDMA-induced behavioral and neuroinflammatory response in mice. Psychopharmacology 2013, 226, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Marchalant, Y.; Wenk, G.L. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neurosci. Lett. 2010, 480, 97–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonsalla, P.K.; Wong, L.Y.; Harris, S.L.; Richardson, J.R.; Khobahy, I.; Li, W.; Gadad, B.S.; German, D.C. Delayed caffeine treatment prevents nigral dopamine neuron loss in a progressive rat model of Parkinson’s disease. Exp. Neurol. 2012, 234, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Dai, S.; An, J.; Li, P.; Chen, X.; Xiong, R.; Liu, P.; Zhao, Y.; Zhu, M.; Liu, X.; et al. Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience 2008, 151, 1198–1207. [Google Scholar] [CrossRef]
- Khairnar, A.; Plumitallo, A.; Frau, L.; Schintu, N.; Morelli, M. Caffeine Enhances Astroglia and Microglia Reactivity Induced by 3,4-Methylenedioxymethamphetamine (‘Ecstasy’) in Mouse Brain. Neurotox. Res. 2010, 17, 435–439. [Google Scholar] [CrossRef]
- Frau, L.; Costa, G.; Porceddu, P.F.; Khairnar, A.; Castelli, M.P.; Ennas, M.G.; Madeddu, C.; Wardas, J.; Morelli, M. Influence of caffeine on 3,4-methylenedioxymethamphetamine-induced dopaminergic neuron degeneration and neuroinflammation is age-dependent. J. Neurochem. 2016, 136, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Madeira, M.H.; Ortin-Martinez, A.; Nadal-Nícolas, F.; Ambrósio, A.F.; Vidal-Sanz, M.; Agudo-Barriuso, M.; Santiago, A.R. Caffeine administration prevents retinal neuroinflammation and loss of retinal ganglion cells in an animal model of glaucoma. Sci. Rep. 2016, 6, 27532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boia, R.; Elvas, F.; Madeira, M.H.; Aires, I.D.; Rodrigues-Neves, A.C.; Tralhão, P.; Szabò, E.C.; Baqi, Y.; Müller, C.E.; Tomè, A.R. Treatment with A2A receptor antagonist KW6002 and caffeine intake regulate microglia reactivity and protect retina against transient ischemic damage. Cell Death Dis. 2017, 8, e3065. [Google Scholar] [CrossRef]
- Hu, J.; Cheng, Y.; Chen, P.; Huang, Z.; Yang, L. Caffeine citrate protects against Sepsis-Associated Encephalopathy and Inhibits the UCP2/NLRP3 Axis in Astrocytes. J. Cytokine Res. 2022, 42, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yu, X.; Zhang, Y.; Liu, N.; Xue, X.; Fu, J. Caffeine treatment started before injury reduces hypoxic–ischemic white-matter damage in neonatal rats by regulating phenotypic microglia polarization. Pediatr. Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ghribi, O.; Geiger, J.D. Caffeine protects against disruptions of the blood-brain barrier in animal models of Alzheimer’s and Parkinson’s diseases. J. Alzheimers Dis. 2010, 20, S127–S141. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.J.; Gawryluk, W.; Wagener, J.F.; Ghribi, O.; Geiger, J.D. Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer’s disease. J. Neuroinflamm. 2008, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Lan, X.; Roche, I.; Liu, R.; Geiger, J.D. Caffeine protects against MPTP-induced blood brain barrier dysfunction in mouse striatum. J. Neurochem. 2008, 107, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Londzin, P.; Zamora, M.; Beata Kakol, B.; Taborek, A.; Folwarczna, J. Potential of Caffeine in Alzheimer’s Disease—A Review of Experimental Studies. Nutrients 2021, 13, 537. [Google Scholar] [CrossRef]
- Asher, S.; Priefer, R. Alzheimer’s disease failed clinical trials. Life Sci. 2022, 306, 120861. [Google Scholar] [CrossRef]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: A review. 3 Biotech. 2022, 12, 55. [Google Scholar] [CrossRef]
- Sharma, B.; kalita, S.; Paul, A.; Mandal, B.; Paul, S. The role of caffeine as an inhibitor in the aggregation of amyloid forming peptides: A unified molecular dynamics simulation and experimental study. RSC Adv. 2016, 6, 78548–78558. [Google Scholar] [CrossRef]
- Mancini, R.S.; Wang, Y.; Weaver, D.F. Phenylindanes in Brewed Coffee Inhibit Amyloid-Beta and Tau Aggregation. Front. Neurosci. 2018, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.S.; Botton, P.H.; Mioranzza, S.; Souza, D.O.; Porciúncula, L.O. Caffeine prevents age-associated recognition memory decline and changes brain-derived neurotrophic factor and tirosine kinase receptor (TrkB) content in mice. Neuroscience 2008, 153, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 42, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Mori, T.; Cao, C.; Mamcarz, M.; Runfeldt, M.; Dickson, A.; Rezai-Zadeh, K.; Tane, J.; Citron, B.A.; Lin, X.; et al. Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J. Alzheimers Dis. 2009, 17, 661–680. [Google Scholar] [CrossRef] [Green Version]
- Dall’Igna, O.P.; Paulo Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and adenosine A2a receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef]
- Quarta, D.; Ferré, S.; Solinas, M.; You, Z.B.; Hockemeyer, J.; Popoli, P.; Goldberg, S.R. Opposite modulatory roles for adenosine A1 and A2A receptors on glutamate and dopamine release in the shell of the nucleus accumbens. Effects of chronic caffeine exposure. J. Neurochem. 2004, 88, 1151–1158. [Google Scholar] [CrossRef]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; a Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciúncula, L.O. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J Alzheimers Dis. 2013, 34, 509–518. [Google Scholar] [CrossRef]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimers Dis. 2010, 1, S167–S174. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Díaz, A.G.; García-Sánchez, A.; Cardona-Muñoz, E.G. Foods with Potential Prooxidant and Antioxidant Effects Involved in Parkinson’s Disease. Oxidative Med. Cell. Longev. 2020, 2020, 6281454. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Pegueroles, J.; Herrmann, A.G.; Henstridge, C.M.; Muñoz, L.; Querol-Vilaseca, M.; Martìn-Paniello, C.S.; Luque-Cabecerans, J.; Clarimon, J.; Belbin, O.; et al. Synaptic phosphorylated α-synuclein in dementia with Lewy bodies. Brain 2017, 140, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Váradi, C. Clinical Features of Parkinson’s Disease: The Evolution of Critical Symptoms. Biology 2020, 9, 103. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Riederer, P. The Nigral Coup in Parkinson’s Disease by α-Synuclein and Its Associated Rebels. Cells 2021, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Percário, S.; da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; de Nazaré Araújo Moreira, T.; Dolabela, M.F. Oxidative Stress in Parkinson’s Disease: Potential Benefits of Antioxidant Supplementation. Oxidative Med. Cell. Longev. 2020, 2020, 2360872. [Google Scholar] [CrossRef]
- Chang, K.H.; Chen, C.M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Park, T.J.; Ali, T.; Kim, M.O. Antioxidant and Neuroprotective Effects of Caffeine against Alzheimer’s and Parkinson’s Disease: Insight into the Role of Nrf-2 and A2AR Signaling. Antioxidants 2020, 9, 902. [Google Scholar] [CrossRef]
- Schepici, G.; Silvestro, S.; Bramanti, P.; Mazzon, E. Caffeine: An Overview of Its Beneficial Effects in Experimental Models and Clinical Trials of Parkinson’s Disease. Mol. Sci. 2020, 21, 4766. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Xie, S.P.; Saw, W.T.; Ho, P.G.H.; Wang, H.; Lei, Z.; Yi, Z.; Tan, E.K. The Therapeutic Implications of Tea Polyphenols Against Dopamine (DA) Neuron Degeneration in Parkinson’s Disease (PD). Cells 2019, 8, 911. [Google Scholar] [CrossRef] [PubMed]
- Machado-Filho, J.A.; Correia, A.O.; Montenegro, A.B.; Nobre, M.E.; Cerqueira, G.S.; Neves, K.R.; da Graça Naffah-Mazzacoratti, M.; Cavalheiro, E.A.; de Castro Brito, G.A.; de Barros Viana, G.S. Caffeine neuroprotective effects on 6-OHDA-lesioned rats are mediated by several factors, including pro-inflammatory cytokines and histone deacetylase inhibitions. Behav. Brain Res. 2014, 264, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.; Schwarzschild, M.A. Adenosine A2A receptor gene disruption protects in an α-synuclein model of Parkinson’s disease. Ann. Neurol. 2012, 71, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.G.; Batalha, V.L.; Miranda, V.H.; Coelho, J.E.; Gomes, R.; Goncalves, F.Q.; Real, J.; Rino, J.; Albino-Teixeira, A.; Cunha, R.A.; et al. Adenosine A2A receptors modulate alphasynuclein aggregation and toxicity. Cereb. Cortex 2017, 27, 718–730. [Google Scholar] [PubMed] [Green Version]
- Ferreira, D.G.; Temido-Ferreira, M.; Vicente Miranda, H.; Batalha, V.L.; Coelho, J.E.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.V.; Rhee, J.S.; Schmitz, M.; et al. Alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 2, 1569–1579. [Google Scholar] [CrossRef]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Romenets, S.R.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postuma, R.B.; Anang, J.; Pelletier, A.; Joseph, L.; Moscovich, M.; Grimes, D.; Furtado, S.; Munhoz, R.P.; Appel-Cresswell, S.; Moro, A.; et al. Caffeine as symptomatic treatment for Parkinson disease (Café-PD): A randomized trial. Neurology 2017, 89, 1795–1803. [Google Scholar] [CrossRef]
- Ascherio, A.; Zhang, S.M.; Herna, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef]
- Palacios, N.; Gao, X.; McCullough, M.L.; Schwarzschild, M.A.; Shah, R.; Gapstur, S.; Asherio, A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov. Disord. 2012, 27, 1276–1282. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, R.; Macklin, E.A.; Hung, A.Y.; Hayes, M.T.; Hyman, B.T.; Wills, A.M.; Gomperts, S.N.; Growdon, J.H.; Ascherio, A.; Scherzer, C.R.; et al. Associations of Lower Caffeine Intake and Plasma Urate Levels with Idiopathic Parkinson’s Disease in the Harvard Biomarkers Study. J. Park. Dis. 2020, 10, 505–510. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. J. Am. Med. Assoc. 2000, 283, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Ebers, G.C. Environmental factors and multiple sclerosis. Lancet Neurol. 2008, 7, 268–277. [Google Scholar] [CrossRef]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [PubMed] [Green Version]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afief, A.R.; Irham, L.M.; Adikusuma, W.; Perwitasari, D.A.; Brahmadhi, A.; Cheung, R. Integration of genomic variants and bioinformatic-based approach to drive drug repurposing for multiple sclerosis. Biochem. Biophys. Rep. 2022, 32, 101337. [Google Scholar] [CrossRef] [PubMed]
- Sharif, K.; Watad, A.; Bragazzi, N.L.; Adawi, M.; Amital, H.; Shoenfeld, Y. Coffee and autoimmunity: More than a mere hot beverage! Autoimmun. Rev. 2017, 16, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Lakin, L.; Davis, B.E.; Binns, C.C.; Currie, K.M.; Rensel, M.R. Comprehensive Approach to Management of Multiple Sclerosis: Addressing Invisible Symptoms—A Narrative Review. Neurol. Ther. 2021, 10, 75–98. [Google Scholar] [CrossRef] [PubMed]
- D’hooghe, M.B.; Haentjens, P.; Nagels, G.; Keyser, J. Alcohol, coffee, fish, smoking and disease progression in multiple sclerosis. Eur. J. Neurol. 2012, 19, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Chen, Y.Y.; Wang, X.S.; Wu, S.Z.; Yang, H.M.; Xu, H.Q.; He, J.C.; Wang, X.T.; Chen, J.F.; Zheng, R.Y. Chronic caffeine treatment attenuates experimental autoimmune encephalomyelitis induced by guinea pig spinal cord homogenates in wistar rats. Brain Res. 2010, 1309, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, S.; Schnermann, J.; Noorbakhsh, F.; Henry, S.; Yong, V.W.; Winston, B.W.; Warren, K.; Power, C. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J. Neurosci. 2004, 24, 1521–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedström, A.K.; Mowry, E.M.; Gianfrancesco, M.A.; Shao, X.; Schaefer, C.A.; Shen, L.; Olsson, T.; Barcellos, L.F.; Alfredsson, L. High consumption of coffee is associated with decreased multiple sclerosis risk; results from two independent studies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Herden, L.; Weissert, R. The Effect of Coffee and Caffeine Consumption on Patients with Multiple Sclerosis-Related Fatigue. Nutrients 2020, 12, 2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Song, K.Y.; Feng, J.Z.; Huang, S.Y.; Guo, X.M.; Zhang, L.; Zhang, G.; Huo, Y.C.; Zhang, R.R.; Ma, Y.; et al. Caffeine Inhibits Activation of the NLRP3 Inflammasome via Autophagy to Attenuate Microglia-Mediated Neuroinflammation in Experimental Autoimmune Encephalomyelitis. J. Mol. Neurosci. 2022, 72, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Keon, M.; Musrie, B.; Dinger, M.; Brennan, S.E.; Santos, J.; Saksena, N.K. Destination Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 596006. [Google Scholar] [CrossRef] [PubMed]
- Hemerková, P.; Vališ, M. Role of Oxidative Stress in the Pathogenesis of Amyotrophic Lateral Sclerosis: Antioxidant Metalloenzymes and Therapeutic Strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 16, 171. [Google Scholar] [CrossRef] [Green Version]
- Leon, D.; Albasanz, J.L.; Ruiz, M.A.; Iglesias, I.; Martin, M. Effect of chronic gestational treatment with caffeine or theophylline on Group I metabotropic glutamate receptors in maternal and fetal brain. J. Neurochem. 2005, 94, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.K.; Higashimori, H.; Tolman, M.; Yang, Y. Suppression of adenosine 2a receptor (A2aR)-mediated adenosine signaling improves disease phenotypes in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2015, 267, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fondell, E.; O’Reilly, É.I.; Fitzgerald, K.C.; Falcone, G.J.; Kolonel, L.N.; Park, Y.; Gapstur, S.M.; Ascherio, A. Intakes of caffeine, coffee and tea and risk of amyotrophic lateral sclerosis: Results from five cohort studies. Amyotroph. Lateral Scler. Front. Degener. 2014, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonin, C.; Duru, C.; Salleron, J.; Hincker, P.; Charles, P.; Delval, A.; Youssov, K.; Burnouf, S.; Azulay, J.P.; Verny, C.; et al. Association between caffeine intake and age at onset in Huntington’s disease. Neurobiol. Dis. 2013, 58, 179–182. [Google Scholar] [CrossRef]
- Koch, E.T.; Raymond, L.A. Dysfunctional striatal dopamine signaling in Huntington’s disease. J. Neurosci. Res. 2019, 97, 1636–1654. [Google Scholar] [CrossRef] [PubMed]
- Popoli, P.; Pintor, A.; Domenici, M.R.; Frank, C.; Tebano, M.T.; Pèzzola, A.; Scarchilli, L.; Quarta, D.; Reggio, R.; Malchiodi-Albedi, F.; et al. Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic acid-induced excitotoxicity: Possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J. Neurosci. 2002, 22, 1967–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, J.; Kumar, A. Improvement of mitochondrial NAD (+)/FAD (+)-linked state-3 respiration by caffeine attenuates quinolinic acid induced motor impairment in rats: Implications in Huntington’s disease. Pharmacol. Rep. 2014, 66, 1148–1155. [Google Scholar] [CrossRef]
- Blum, D.; Chern, Y.; Domenici, M.R.; Buée, L.; Lin, C.Y.; Rea, W.; Ferré, S.; Popoli, P. The Role of Adenosine Tone and Adenosine Receptors in Huntington’s Disease. J. Caffeine Adenosine Res. 2018, 8, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Nehlig, A. Interindividual Differences in Caffeine Metabolism and Factors Driving Caffeine Consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faudone, G.; Arifi, S.; Merk, D. The Medicinal Chemistry of Caffeine. J. Med. Chem. 2021, 64, 7156–7178. [Google Scholar] [CrossRef]
- Mastropietro, D.; Park, K.; Omidian, H. Polymers in Oral Drug Delivery. Fac. Books Book Chapters 2017, 17, 430–444. [Google Scholar]
- Kolbina, M.; Bodmeier, R.; Körber, M. Saturated phosphatidylcholine as matrix former for oral extended release dosage forms. Eur. J. Pharm. Sci. 2017, 108, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.; Wu, L.; Zhang, C.; Davis, R.M.; Xu, B. Natural product-based nanomedicine: Recent advances and issues. Int. J. Nanomed. 2015, 10, 6055–6074. [Google Scholar]
- Loquercio, A.; Castell-Perez, E.; Gomes, C.; Moreira, R.G. Preparation of Chitosan-Alginate Nanoparticles for Trans-cinnamaldehyde Entrapment. J. Food Sci. 2015, 80, N2305–N2315. [Google Scholar] [CrossRef]
- Milkova, V.; Goycoolea, F.M. Encapsulation of caffeine in polysaccharide oil-core nanocapsules. Colloid Polym. Sci. 2020, 298, 1035–1041. [Google Scholar] [CrossRef]
- Fonseca, L.R.; Santos, T.P.; Czaikoski, A.; Cunha, R.L. Microfluidics-based production of chitosan-gellan nanocomplexes encapsulating caffeine. Food Res. Int. 2022, 151, 110885. [Google Scholar] [CrossRef] [PubMed]
- Noor, N.; Shah, A.; Gani, A.; Gani, A.; Masoodi, F.A. Microencapsulation of caffeine loaded in polysaccharide based delivery system. Food Hydrocoll. 2018, 82, 312–321. [Google Scholar] [CrossRef]
- Massella, D.; Celasco, E.; Salaün, F.; Ferri, A.; Barresi, A.A. Overcoming the Limits of Flash Nanoprecipitation: Effective Loading of Hydrophilic Drug into Polymeric Nanoparticles with Controlled Structure. Polymer 2018, 10, 1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Bourbon, A.I.; Cerqueira, M.A.; Vicente, A.A. Encapsulation and controlled release of bioactive compounds in lactoferrin-glycomacropeptide nanohydrogels: Curcumin and caffeine as model compounds. J. Food Eng. 2016, 180, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Araújo, D.; Rodrigues, T.; Alves, V.D.; Freitas, F. Chitin-Glucan Complex Hydrogels: Optimization of Gel Formation and Demonstration of Drug Loading and Release Ability. Polymer 2022, 14, 785. [Google Scholar] [CrossRef]
- Artusio, F.; Ferri, A.; Gigante, V.; Massella, D.; Mazzarino, I.; Sangermano, M.; Barresi, A.; Pisano, R. Synthesis of high payload nanohydrogels for the encapsulation of hydrophilic molecules via inverse miniemulsion polymerization: Caffeine as a case study. Drug Dev. Ind. Pharm. 2019, 45, 1862–1870. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruggiero, M.; Calvello, R.; Porro, C.; Messina, G.; Cianciulli, A.; Panaro, M.A. Neurodegenerative Diseases: Can Caffeine Be a Powerful Ally to Weaken Neuroinflammation? Int. J. Mol. Sci. 2022, 23, 12958. https://doi.org/10.3390/ijms232112958
Ruggiero M, Calvello R, Porro C, Messina G, Cianciulli A, Panaro MA. Neurodegenerative Diseases: Can Caffeine Be a Powerful Ally to Weaken Neuroinflammation? International Journal of Molecular Sciences. 2022; 23(21):12958. https://doi.org/10.3390/ijms232112958
Chicago/Turabian StyleRuggiero, Melania, Rosa Calvello, Chiara Porro, Giovanni Messina, Antonia Cianciulli, and Maria Antonietta Panaro. 2022. "Neurodegenerative Diseases: Can Caffeine Be a Powerful Ally to Weaken Neuroinflammation?" International Journal of Molecular Sciences 23, no. 21: 12958. https://doi.org/10.3390/ijms232112958
APA StyleRuggiero, M., Calvello, R., Porro, C., Messina, G., Cianciulli, A., & Panaro, M. A. (2022). Neurodegenerative Diseases: Can Caffeine Be a Powerful Ally to Weaken Neuroinflammation? International Journal of Molecular Sciences, 23(21), 12958. https://doi.org/10.3390/ijms232112958