
Vertebrate Animal Models of RP59: Current Status and Future Prospects

 ,
,  ,
, 
Abstract
:1. Introduction
2. Zebrafish Model of RP59
3. Mouse Models of RP59
3.1. K42E Dhdds Knock-In Mouse
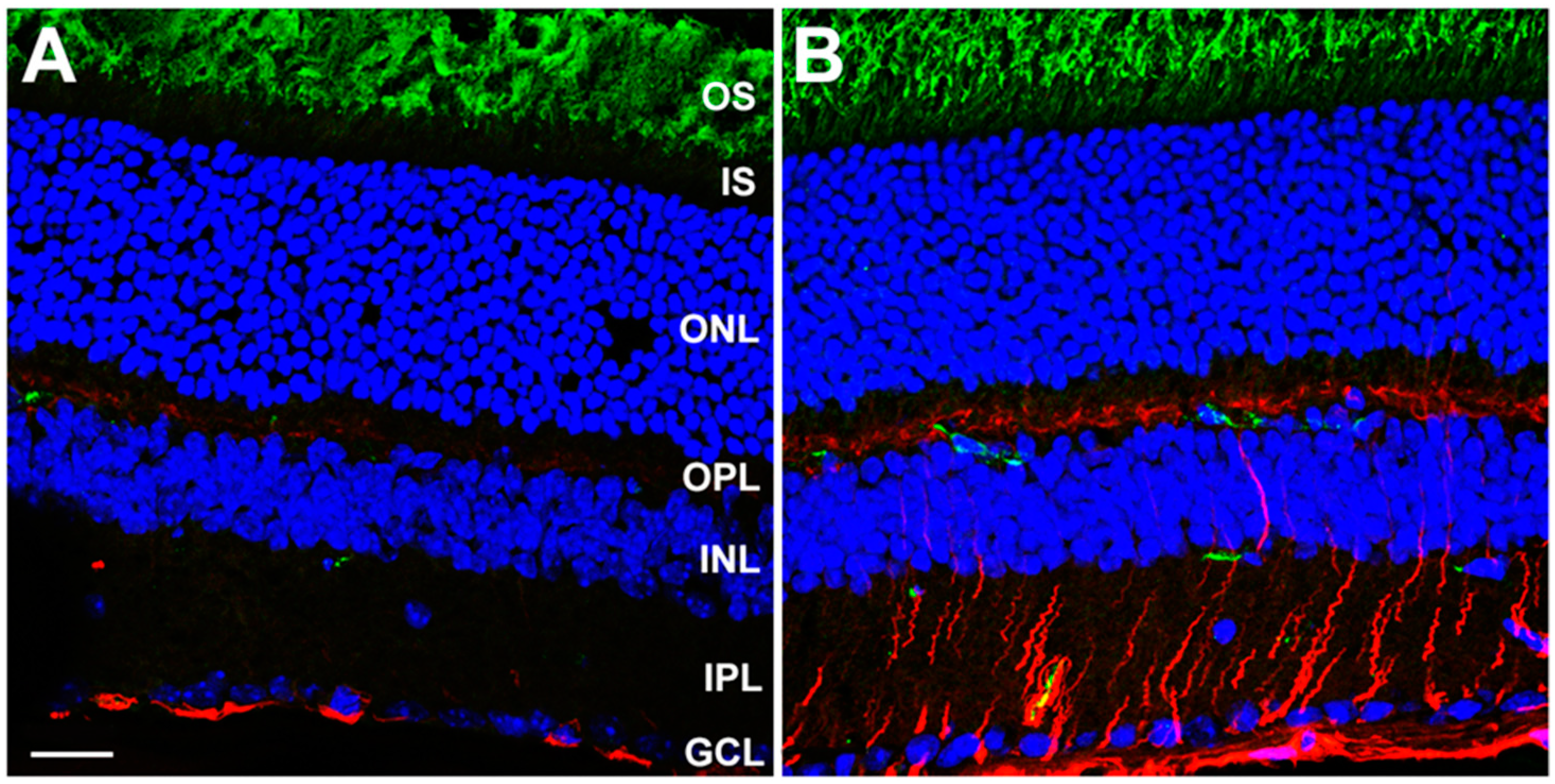
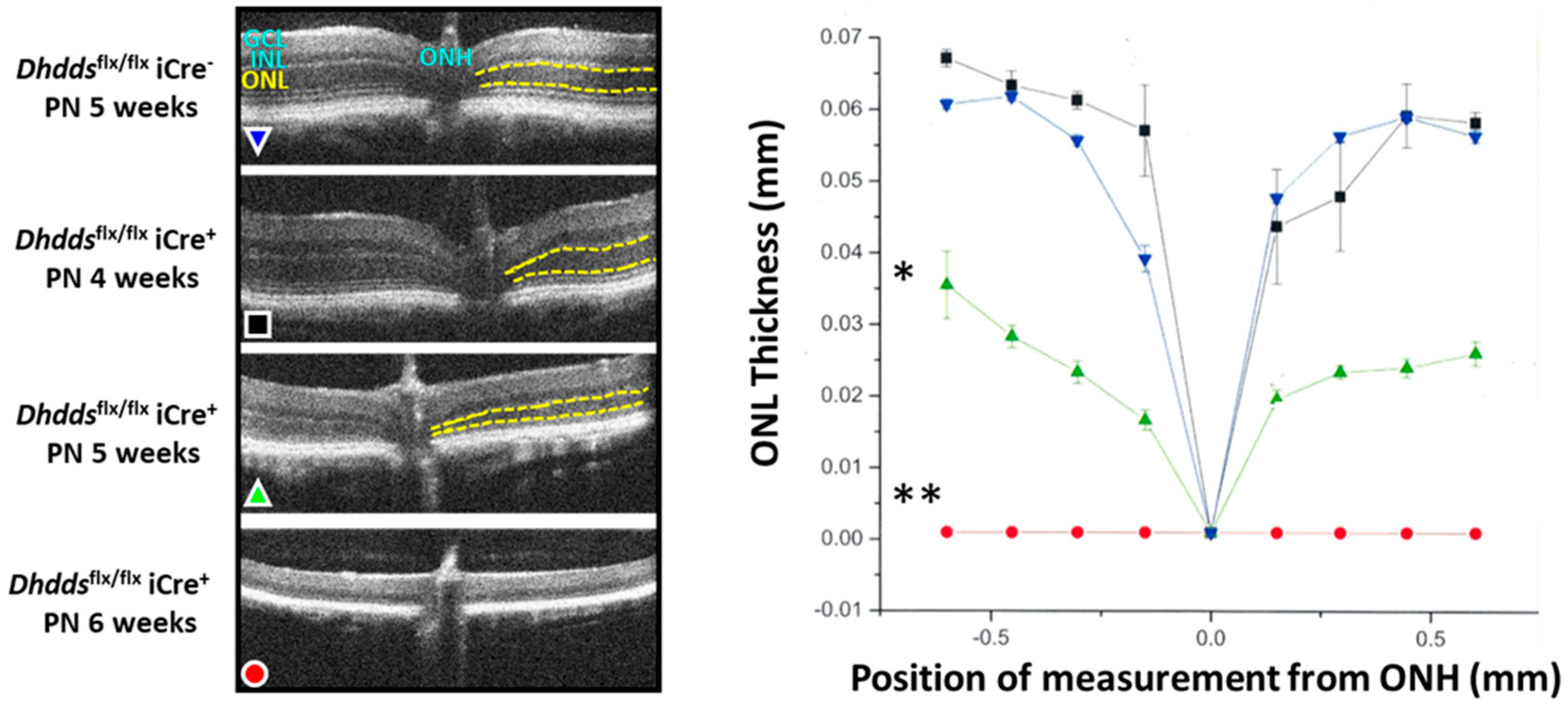
3.2. Rod-Specific Dhdds Knockout Mouse
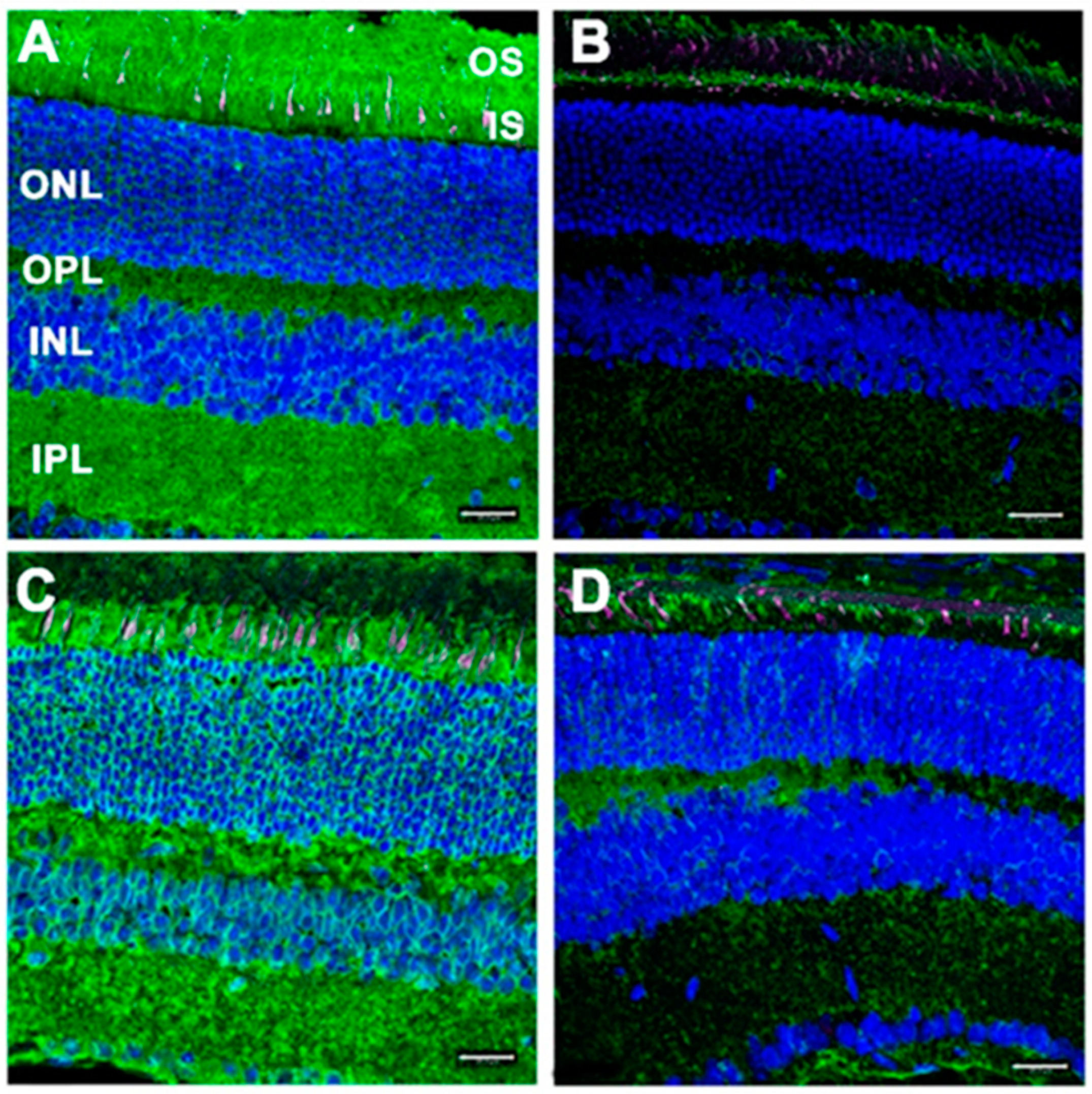
3.3. RPE-Specific Dhdds Knockout Mouse
3.4. Nogo-B Receptor Mutants as RP59 Models
3.5. Emerging New Mouse Models of RP59
3.6. Retinal Degeneration in a Drosophila Dhdds Knockdown Model
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Züchner, S.; Dallman, J.; Wen, R.; Beecham, G.; Naj, A.; Farooq, A.; Kohli, M.A.; Whitehead, P.L.; Hulme, W.; Konidari, I.; et al. Whole-Exome Sequencing Links a Variant in DHDDS to Retinitis Pigmentosa. Am. J. Hum. Genet. 2011, 88, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Zelinger, L.; Banin, E.; Obolensky, A.; Mizrahi-Meissonnier, L.; Beryozkin, A.; Bandah-Rozenfeld, D.; Frenkel, S.; Ben-Yosef, T.; Merin, S.; Schwartz, S.B.; et al. A Missense Mutation in DHDDS, Encoding Dehydrodolichyl Diphosphate Synthase, Is Associated with Autosomal-Recessive Retinitis Pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 2011, 88, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimchi, A.; Khateb, S.; Wen, R.; Guan, Z.; Obolensky, A.; Beryozkin, A.; Kurtzman, S.; Blumenfeld, A.; Pras, E.; Jacobson, S.G.; et al. Nonsyndromic Retinitis Pigmentosa in the Ashkenazi Jewish Population: Genetic and Clinical Aspects. Ophthalmology 2018, 125, 725–734. [Google Scholar] [CrossRef]
- Biswas, P.; Duncan, J.L.; Maranhao, B.; Kozak, I.; Branham, K.; Gabriel, L.; Lin, J.H.; Barteselli, G.; Navani, M.; Suk, J.; et al. Genetic analysis of 10 pedigrees with inherited retinal degeneration by exome sequencing and phenotype-genotype association. Physiol. Genom. 2017, 49, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Sabry, S.; Vuillaumier-Barrot, S.; Mintet, E.; Fasseu, M.; Valayannopoulos, V.; Héron, D.; Dorison, N.; Mignot, C.; Seta, N.; Chantret, I.; et al. A case of fatal Type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J. Rare Dis. 2016, 11, 84. [Google Scholar] [CrossRef] [Green Version]
- Wen, R.; Lam, B.L.; Guan, Z. Aberrant dolichol chain lengths as biomarkers for retinitis pigmentosa caused by impaired dolichol biosynthesis. J. Lipid Res. 2013, 54, 3516–3522. [Google Scholar] [CrossRef] [Green Version]
- Lam, B.L.; Züchner, S.L.; Dallman, J.; Wen, R.; Alfonso, E.C.; Vance, J.; Peričak-Vance, M.A. Mutation K42E in Dehydrodolichol Diphosphate Synthase (DHDDS) Causes Recessive Retinitis Pigmentosa. In Retinal Degenerative Diseases; Springer: New York, NY, USA, 2014; Volume 801, pp. 165–170. [Google Scholar] [CrossRef]
- Venturini, G.; Koskiniemi-Kuendig, H.; Harper, S.; Berson, E.L.; Rivolta, C. Two specific mutations are prevalent causes of recessive retinitis pigmentosa in North American patients of Jewish ancestry. Genet. Med. 2015, 17, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Hariri, A.H.; Gui, W.; Datoo O’Keefe, G.A.; Ip, M.S.; Sadda, S.R.; Gorin, M.B. Ultra-Widefield Fundus Autofluorescence Imaging of Patients with Retinitis Pigmentosa: A Standardized Grading System in Different Genotypes. Ophthalmol. Retina 2018, 2, 735–745. [Google Scholar] [CrossRef]
- Brasher, M.I.; Surmacz, L.; Leong, B.; Pitcher, J.; Swiezewska, E.; Pichersky, E.; Akhtar, T.A. A two-component enzyme complex is required for dolichol biosynthesis in tomato. Plant J. 2015, 82, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Sato, K.; Nishikawa, S.-I.; Hirata, A.; Kato, J.-I.; Nakano, A. The Yeast RER2 Gene, Identified by Endoplasmic Reticulum Protein Localization Mutations, Encodes cis-Prenyltransferase, a Key Enzyme in Dolichol Synthesis. Mol. Cell. Biol. 1999, 19, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-El, M.L.; Vaňková, P.; Yeheskel, A.; Simhaev, L.; Engel, H.; Man, P.; Haitin, Y.; Giladi, M. Structural basis of heterotetrameric assembly and disease mutations in the human cis-prenyltransferase complex. Nat. Commun. 2020, 11, 5273. [Google Scholar] [CrossRef] [PubMed]
- Edani, B.H.; Grabińska, K.A.; Zhang, R.; Park, E.J.; Siciliano, B.; Surmacz, L.; Ha, Y.; Sessa, W.C. Structural elucidation of the cis -prenyltransferase NgBR/DHDDS complex reveals insights in regulation of protein glycosylation. Proc. Natl. Acad. Sci. USA 2020, 117, 20794–20802. [Google Scholar] [CrossRef]
- Orlean, P. Enzymes that recognize dolichols participate in three glycosylation pathways and are required for protein secretion. Biochem. Cell Biol. 1992, 70, 438–447. [Google Scholar] [CrossRef]
- Doucey, M.-A.; Hess, D.; Cacan, R.; Hofsteenge, J. Protein C-Mannosylation Is Enzyme-catalysed and Uses Dolichyl-Phosphate-Mannose as a Precursor. Mol. Biol. Cell 1998, 9, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, J.; Kinoshita, T. GPI-anchor biosynthesis. Trends Biochem. Sci. 1995, 20, 367–371. [Google Scholar] [CrossRef]
- Kornfeld, R.; Kornfeld, S. Assembly of Asparagine-Linked Oligosaccharides. Annu. Rev. Biochem. 1985, 54, 631–664. [Google Scholar] [CrossRef] [PubMed]
- Welti, M. Regulation of dolichol-linked glycosylation. Glycoconj. J. 2012, 30, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Kean, E.L. Studies on the activation by dolichol-P-mannose of the biosynthesis of GlcNAc-P-P-dolichol and the topography of the GlcNAc-transferases concerned with the synthesis of GlcNAc-P-P-dolichol and (GlcNAc)2-P-P-dolichol: A review. Biochem. Cell Biol. 1992, 70, 413–421. [Google Scholar] [CrossRef]
- Kean, E.L. The dolichol pathway in the retina and its involvement in the glycosylation of rhodopsin. Biochim. Biophys. Acta 1999, 1473, 272–285. [Google Scholar] [CrossRef]
- Plantner, J.J.; Kean, E.L. The dolichol pathway in the retina: Oligosaccharide-lipid biosynthesis. Exp. Eye Res. 1988, 46, 785–800. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Rapp, L.M.; Hollyfield, J.G. Photoreceptor-specific degeneration caused by tunicamycin. Nature 1984, 311, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.H.; Williams, D.S.; Neitz, J.; Fariss, R.N.; Fliesler, S.J. Tunicamycin-induced degeneration in cone photoreceptors. Vis. Neurosci. 1988, 1, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J.; Rayborn, E.M.; Hollyfield, J.G. Membrane morphogenesis in retinal rod outer segments: Inhibition by tunicamycin. J. Cell Biol. 1985, 100, 574–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliesler, S.J.; Basinger, S.F. Tunicamycin blocks the incorporation of opsin into retinal rod outer segment membranes. Proc. Natl. Acad. Sci. USA 1985, 82, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Zdrazilova, L.; Kuchar, L.; Ondruskova, N.; Honzik, T.; Hansikova, H. A new role for dolichol isoform profile in the diagnostics of CDG disorders. Clin. Chim. Acta 2020, 507, 88–93. [Google Scholar] [CrossRef]
- Cantagrel, V.; Lefeber, D.J. From glycosylation disorders to dolichol biosynthesis defects: A new class of metabolic diseases. J. Inherit. Metab. Dis. 2011, 34, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.; Gadomski, T.; Kozicz, T.; Morava, E. Congenital disorders of glycosylation: New defects and still counting. J. Inherit. Metab. Dis. 2014, 37, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Cylwik, B.; Naklicki, M.; Chrostek, L.; Gruszewska, E. Congenital Disorders of Glycosylation. Part I. Defects of Protein N-Glycosylation. Acta Biochim. Pol. 2013, 60, 151–161. [Google Scholar] [CrossRef]
- Sparks, S.E.; Krasnewich, D.M. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Jiao, X.; Xue, Y.; Yang, S.; Gong, P.; Niu, Y.; Wang, Q.; Yang, H.; Xiong, H.; Zhang, Y.; Yang, Z. Phenotype of heterozygous variants of dehydrodolichol diphosphate synthase. Dev. Med. Child Neurol. 2022, 64, 125–134. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.J.; Son, H.; Hwang, S.; Kang, M.; Chu, K.; Lee, S.K.; Moon, J. Adult-onset rapidly worsening progressive myoclonic epilepsy caused by a novel variant in DHDDS. Ann. Clin. Transl. Neurol. 2021, 8, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Canafoglia, L.; Franceschetti, S.; Gambardella, A.; Striano, P.; Giallonardo, A.T.; Tinuper, P.; Di Bonaventura, C.; Michelucci, R.; Ferlazzo, E.; Granata, T.; et al. Progressive Myoclonus Epilepsies: Diagnostic Yield with Next-Generation Sequencing in Previously Unsolved Cases. Neurol. Genet. 2021, 7, e641. [Google Scholar] [CrossRef]
- Galosi, S.; Edani, B.H.; Martinelli, S.; Hansikova, H.; Eklund, A.E.; Caputi, C.; Masuelli, L.; Corsten-Janssen, N.; Srour, M.; Oegema, R.; et al. De novo DHDDS variants cause a neurodevelopmental and neurodegenerative disorder with myoclonus. Brain 2021, 145, 208–223. [Google Scholar] [CrossRef]
- Wood, K.; Montgomery, T.; Devlin, A.M. DHDDS related epilepsy—Report of familial cases and review of the literature. Seizure 2021, 91, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Togashi, N.; Fujita, A.; Shibuya, M.; Uneoka, S.; Miyabayashi, T.; Sato, R.; Okubo, Y.; Endo, W.; Inui, T.; Jin, K.; et al. Fifteen-year follow-up of a patient with a DHDDS variant with non-progressive early onset myoclonic tremor and rare generalized epilepsy. Brain Dev. 2020, 42, 696–699. [Google Scholar] [CrossRef]
- Piccolo, G.; Amadori, E.; Vari, M.S.; Marchese, F.; Riva, A.; Ghirotto, V.; Iacomino, M.; Salpietro, V.; Zara, F.; Striano, P. Complex Neurological Phenotype Associated with a De Novo DHDDS Mutation in a Boy with Intellectual Disability, Refractory Epilepsy, and Movement Disorder. J. Pediatr. Genet. 2021, 10, 236–238. [Google Scholar] [CrossRef]
- Kim, J.; Kim, I.; Koh, S.-B. A novel variant of dehydrodolichol diphosphate synthase (DHDDS) mutation with adult-onset progressive myoclonus ataxia. Park. Relat. Disord. 2021, 87, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Courage, C.; Oliver, K.L.; Park, E.J.; Cameron, J.M.; Grabińska, K.A.; Muona, M.; Canafoglia, L.; Gambardella, A.; Said, E.; Afawi, Z.; et al. Progressive myoclonus epilepsies—Residual unsolved cases have marked genetic heterogeneity including dolichol-dependent protein glycosylation pathway genes. Am. J. Hum. Genet. 2021, 108, 722–738. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-Syndromic Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Noel, N.C.L.; MacDonald, I.M.; Allison, W.T. Zebrafish Models of Photoreceptor Dysfunction and Degeneration. Biomolecules 2021, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Angueyra, J.M.; Kindt, K.S. Leveraging Zebrafish to Study Retinal Degenerations. Front. Cell Dev. Biol. 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Wen, R.; Dallman, J.E.; Li, Y.; Züchner, S.L.; Vance, J.M.; Peričak-Vance, M.A.; Lam, B.L. Knock-Down DHDDS Expression Induces Photoreceptor Degeneration in Zebrafish. Adv. Exp. Med. Biol. 2014, 801, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.V.; Hageman, G.S.; Blanks, J.C. Interphotoreceptor matrix domains ensheath vertebrate cone photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 1986, 27, 129–135. [Google Scholar]
- Johnson, L.V.; Hageman, G.S. Structural and compositional analyses of isolated cone matrix sheaths. Investig. Ophthalmol. Vis. Sci. 1991, 32, 1951–1957. [Google Scholar]
- Collin, G.B.; Gogna, N.; Chang, B.; Damkham, N.; Pinkney, J.; Hyde, L.F.; Stone, L.; Naggert, J.K.; Nishina, P.M.; Krebs, M.P. Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss. Cells 2020, 9, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budzynski, E.; Lee, Y.; Sakamoto, K.; Naggert, J.K.; Nishina, P.M. From Vivarium to Bedside: Lessons Learned from Animal Models. Ophthalmic Genet. 2006, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schaffer, A.A.; Yu, Y.-K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef]
- Rao, S.R.; Fliesler, S.J.; Kotla, P.; Nguyen, M.N.; Pittler, S.J. Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59. Cells 2020, 9, 896. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Ignatova, I.; Nguyen, M.N.; Cobb, P.I.; Rao, S.R.; Bocchero, U.; Fliesler, S.J.; Pahlberg, J.; Pittler, S.J. A K42e Knockin Mouse Model of Rp59 Exhibits a Negative Erg and Defective Postsynaptic Signal Transmission. Invest. Ophthamol. Vis. Sci. 2021, 62, 2958. [Google Scholar]
- Pittler, S.J.; Rao, S.R.; Fliesler, S.J.; Messinger, S.J.; Sherry, D.M. Ultrastructural Changes in the RPE and Outer Retina in a Mouse Knock-in Model of RP59. Invest. Ophthalmol. Vis. Sci. 2022, 63, 2377. [Google Scholar]
- Li, S.; Chen, D.; Sauve, Y.; McCandless, J.; Chen, Y.J.; Chen, C.K. Rhodopsin-iCre Transgenic Mouse Line for Cre-Mediated Rod-Specific Gene Targeting. Genesis 2005, 41, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.R.; Skelton, L.A.; Wu, F.; Onysk, A.; Spolnik, G.; Danikiewicz, W.; Butler, M.C.; Stacks, D.A.; Surmacz, L.; Mu, X.; et al. Retinal Degeneration Caused by Rod-Specific Dhdds Ablation Occurs without Concomitant Inhibition of Protein N-Glycosylation. iScience 2020, 23, 101198. [Google Scholar] [CrossRef]
- Murray, A.R.; Fliesler, S.J.; Al-Ubaidi, M.R. Rhodopsin: The Functional Significance of Asn-Linked Glycosylation and Other Post-Translational Modifications. Ophthalmic Genet. 2009, 30, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winchester, B.G. Lysosomal Membrane Proteins. Eur. J. Paediatr. Neurol. 2001, 5 (Suppl. A), 11–19. [Google Scholar] [CrossRef]
- DeRamus, M.L.; Davis, S.J.; Rao, S.R.; Nyankerh, C.; Stacks, D.; Kraft, T.W.; Fliesler, S.J.; Pittler, S.J. Selective Ablation of Dehydrodolichyl Diphosphate Synthase in Murine Retinal Pigment Epithelium (RPE) Causes RPE Atrophy and Retinal Degeneration. Cells 2020, 9, 771. [Google Scholar] [CrossRef] [Green Version]
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Grabińska, K.A.; Guan, Z.; Stránecký, V.; Hartmannová, H.; Hodaňová, K.; Barešová, V.; Sovová, J.; Jozsef, L.; Ondrušková, N.; et al. Mutation of Nogo-B Receptor, a Subunit of cis-Prenyltransferase, Causes a Congenital Disorder of Glycosylation. Cell Metab. 2014, 20, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.N.; Chakraborty, D.; Cobb, P.I.; Fliesler, S.J.; Pittler, S.J. Generation and Characterization of Novel DHDDS Knock-in Mutation Mouse Models of RP59. Invest. Ophthalmol. Vis. Sci. 2021, 62, 1464. [Google Scholar]
- Brandwine, T.; Ifrah, R.; Bialistoky, T.; Zaguri, R.; Rhodes-Mordov, E.; Mizrahi-Meissonnier, L.; Sharon, D.; Katanaev, V.L.; Gerlitz, O.; Minke, B. Knockdown of Dehydrodolichyl Diphosphate Synthase in the Drosophila Retina Leads to a Unique Pattern of Retinal Degeneration. Front. Mol. Neurosci. 2021, 14, 693967. [Google Scholar] [CrossRef] [PubMed]
- Skoneczny, M.; Kludkiewicz, B.; Swiezewska, E.; Szkopińska, A. Activity of Pichia pastoris alternative cis-prenyltransferase is correlated with proliferation of peroxisomes. Cell Biol. Int. 2006, 30, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Medina-Cano, D.; Ucuncu, E.; Nguyen, L.S.; Nicouleau, M.; Lipecka, J.; Bizot, J.-C.; Thiel, C.; Foulquier, F.; Lefort, N.; Faivre-Sarrailh, C.; et al. High N-glycan multiplicity is critical for neuronal adhesion and sensitizes the developing cerebellum to N-glycosylation defect. eLife 2018, 7, e38309. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Lundberg, E.; Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet. 2022, 247, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2009, 13, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Ambrozkiewicz, M.C.; Benseler, F.; Chen, C.; Dumontier, E.; Falkner, S.; Furlanis, E.; Gomez, A.M.; Hoshina, N.; Huang, W.-H.; et al. Optimizing Nervous System-Specific Gene Targeting with Cre Driver Lines: Prevalence of Germline Recombination and Influencing Factors. Neuron 2020, 106, 37–65.e5. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Biswas [5] | Kimchi [4] | Hariri [10] | Reference |
| DHDDS (p. K42E and p. T206A) | DHDDS (p. K42E, p. T206A and p. R98W) | DHDDS (p. K42E) | Mutation |
| 4 (4) | 30, 28 with K42E (22) | 4 (4) | N (n) |
| AJ | AJ | NR | Ancestry |
| Negative FH | NR | NR | Family History |
| Night blindness started at age 17 y/o in one patient | Major loss of vision during the third and fourth decades | Mean age at diagnosis was 20 y/o (r, 17–22) | Medical/Ocular History |
| At age 22 y/o, 20/25 OU; at age 32 y/o, 20/40; at age 33 y/o, 20/50; at age 40 y/o, 20/70 | NR | Four patients: 20/20 OU; (20/30 OD, 20/40 OS); (20/40 OD, 20/50 OS); (20/60 OD, 20/100 OS) | BCVA |
| Lens, PSC; Fundus, ONH pallor, attenuated retinal blood vessels, pigmentary RPE changes with white spots in the periphery; FAF, NR | Lens, NR; Fundus, atrophy and bone spicule pigmentation which increased in density and involved the macula with age; FAF, perifoveal ring of hyper-autofluorescence | Lens, NR; Fundus, NR; FAF, reduced AF of different grades, abnormal autofluorescence in the macula, complete disc hyper-autofluorescence in two patients | Lens, Fundus, FAF |
| Progressive constriction over time to <20° diameter by age 31 y/o | NR | NR | Visual Field (VF) |
| Scotopic and photopic ERG, severely reduced responses with similar reduction in a and b waves’ amplitudes | Scotopic ERG was non-detectable at first testing; Cone flicker ERG became non-detectable by 28 y/0 | NR | ERG |
| NR | Loss/disruption of the ellipsoid zone, ONL, and RPE; small foveal islands of PRs which ultimately disappeared with age | NR | OCT |
| Chorioretinal biopsy, intact RPE with significant degeneration of all other retinal layers including GCL and PRs (inner and outer segments, and nuclei); Audiogram, bilateral normal hearing function | Color vision, tritanopia in two patients; EOG Arden ratio *** was reduced (r, 100–144%) | NR | Others |
| Venturini [9] | Lam [8] | Reference | |
| DHDDS (p. K42E) | DHDDS (p. K42E) | Mutation | |
| 6 ** (5) | 3 (3) | N (n) | |
| Three of Jewish ancestry, two of mixed ethnicities | AJ | Ancestry | |
| Positive FH | NR | Family History | |
| Night blindness started at 27.8 y/o (r, 21–32) | Night blindness and peripheral vision defects by 15 y/o in two siblings | Medical/Ocular History | |
| Five patients: 20/20 OU; 20/25 OU; 20/30 OU; (20/50 OD, 20/30 OS); (20/100 OD, 20/30 OS) | At diagnosis, from 20/20 to 20/25; in mid-thirties, from 20/40 to 20/400; one patient was LP OU by 30 y/o | BCVA | |
| Lens, PSC in all (one case was pseudophakic OU); Fundus, peripheral bone spicule pigmentation in all, granular macula in 2 patients; FAF, NR | Lens, NR; Fundus, pigmentary retinal degeneration; FAF, NR | Lens, Fundus, FAF | |
| VF loss onset at a mean age of 28.6 y/o; in fourth decade, most VF areas were below 50% of normal | Constricted to <10° at age 36 y/o in two siblings | Visual Field (VF) | |
| Mean amplitude for Scotopic ERG was 21.9 mv (r, 2.6–47) and 19.6 mv (r, 1.4–44.1) for OD and OS, respectively; for photopic ERG was 2.1 mv (r, 0.21–5.9) and 2.01 mv (r, 0.25–5.64) for OD and OS, respectively | Non-detectable in two siblings | ERG | |
| NR | NR | OCT | |
| Dark adaptation, threshold in two patients was 0.5 and 1.5 log units above normal | Plasma transferrin isoelectric focusing gel, all patterns were normal; protein glycosylation was normal | Others | |
| Zelinger [3] | Zuchner [2] | Reference | |
| DHDDS (p. K42E) | DHDDS (p. K42E) | Mutation | |
| 21 (18, one family with longitudinal data) | 3 (3) | N (n) | |
| AJ | AJ | Ancestry | |
| NR | NR | Family History | |
| NR | Lytic bone disease in two siblings; retinitis pigmentosa diagnosis in teenage years | Medical/Ocular History | |
| Ranged from LP to 20/20 (only four eyes with 20/20); 20/200 or worse in two siblings from the family with longitudinal data by age 30–31 y/o | NR | BCVA | |
| Lens, NR; Fundus, waxy ONH; attenuated retinal blood vessels; bone spicule-like pigmentation; FAF, preserved RPE islands corresponding to regions of preserved PRs | Lens, NR; Fundus, pigmentary retinal degeneration; FAF, NR | Lens, Fundus, FAF | |
| Reduced peripheral function; small central islands of vision remaining later in life | NR | Visual Field (VF) | |
| Non-detectable in most patients; borderline rod ERG amplitude in one father’s recording from the family with longitudinal data | Impaired rod and cone responses | ERG | |
| Preserved PRs’ layer in the fovea which declined in thickness away from the fovea; occasional CME; in the family with longitudinal data, PRs were not detectable around the fovea in two siblings, while one sibling had a locus of PRs nasal to the ONH | NR | OCT | |
| Dark Adaptation, progressively diminished until only cone-mediated function was detectable; DHDDS staining, prominent in the basal aspect of RPE cells, IS of the cones, ellipsoid and myoid regions of the rods, weak signal in other retinal layers | Neurologic examination, bone X-ray survey and density scan, brain MRI, echocardiogram, lipid profile, thyroid function studies, serum IGF-binding protein 1 and 2, serum clotting factors, and antithrombin III were normal | Others | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fliesler, S.J.; Ramachandra Rao, S.; Nguyen, M.N.; KhalafAllah, M.T.; Pittler, S.J. Vertebrate Animal Models of RP59: Current Status and Future Prospects. Int. J. Mol. Sci. 2022, 23, 13324. https://doi.org/10.3390/ijms232113324
Fliesler SJ, Ramachandra Rao S, Nguyen MN, KhalafAllah MT, Pittler SJ. Vertebrate Animal Models of RP59: Current Status and Future Prospects. International Journal of Molecular Sciences. 2022; 23(21):13324. https://doi.org/10.3390/ijms232113324
Chicago/Turabian StyleFliesler, Steven J., Sriganesh Ramachandra Rao, Mai N. Nguyen, Mahmoud Tawfik KhalafAllah, and Steven J. Pittler. 2022. "Vertebrate Animal Models of RP59: Current Status and Future Prospects" International Journal of Molecular Sciences 23, no. 21: 13324. https://doi.org/10.3390/ijms232113324
APA StyleFliesler, S. J., Ramachandra Rao, S., Nguyen, M. N., KhalafAllah, M. T., & Pittler, S. J. (2022). Vertebrate Animal Models of RP59: Current Status and Future Prospects. International Journal of Molecular Sciences, 23(21), 13324. https://doi.org/10.3390/ijms232113324