
Cardioprotection by Hypothyroidism Is Not Mediated by Favorable Hemodynamics—Role of Canonical Thyroid Hormone Receptor Alpha Signaling

 , ,
, , 
Abstract
:1. Introduction
2. Results
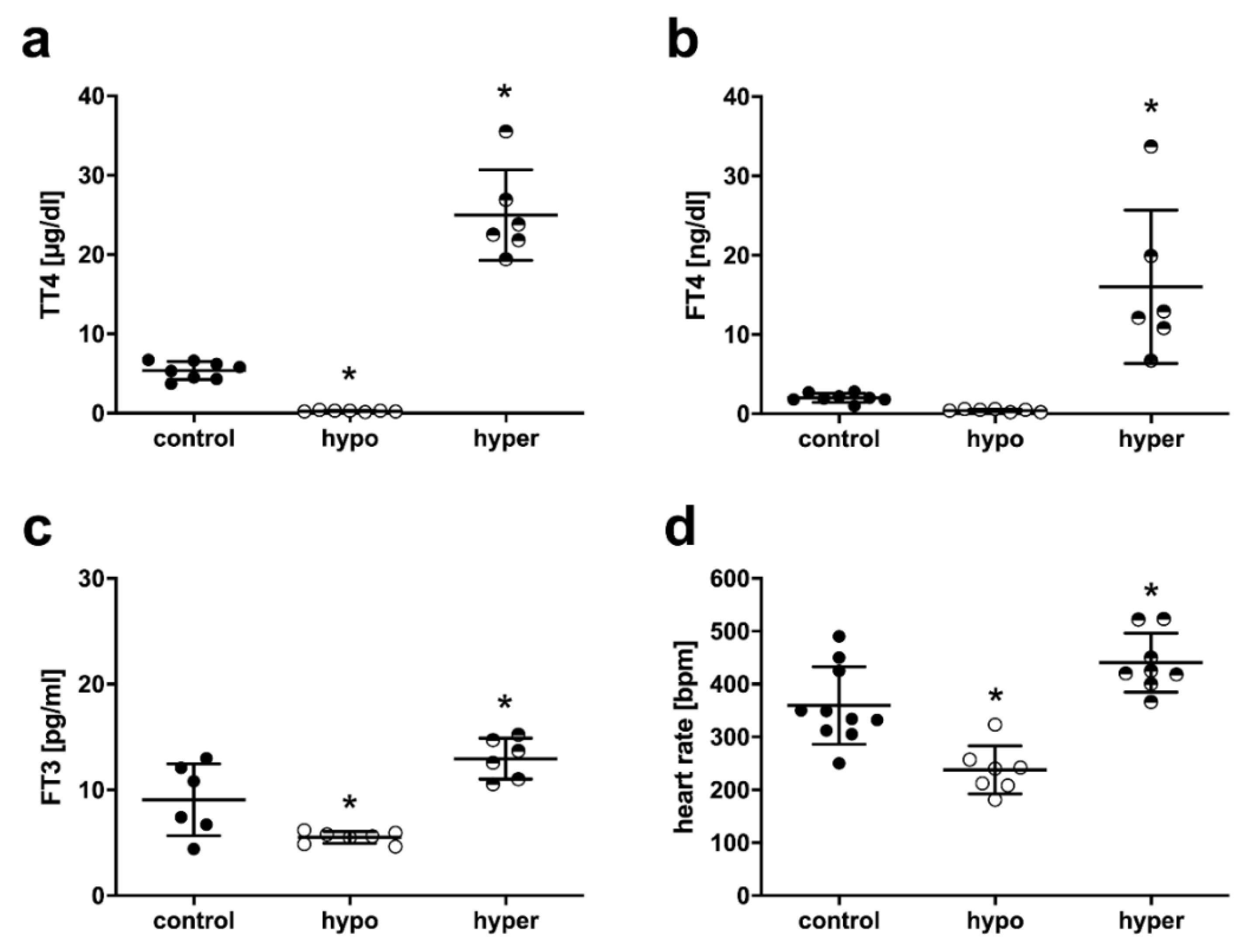
2.1. Serum TH Status and Heart Rate in Mice with Thyroid Dysfunction
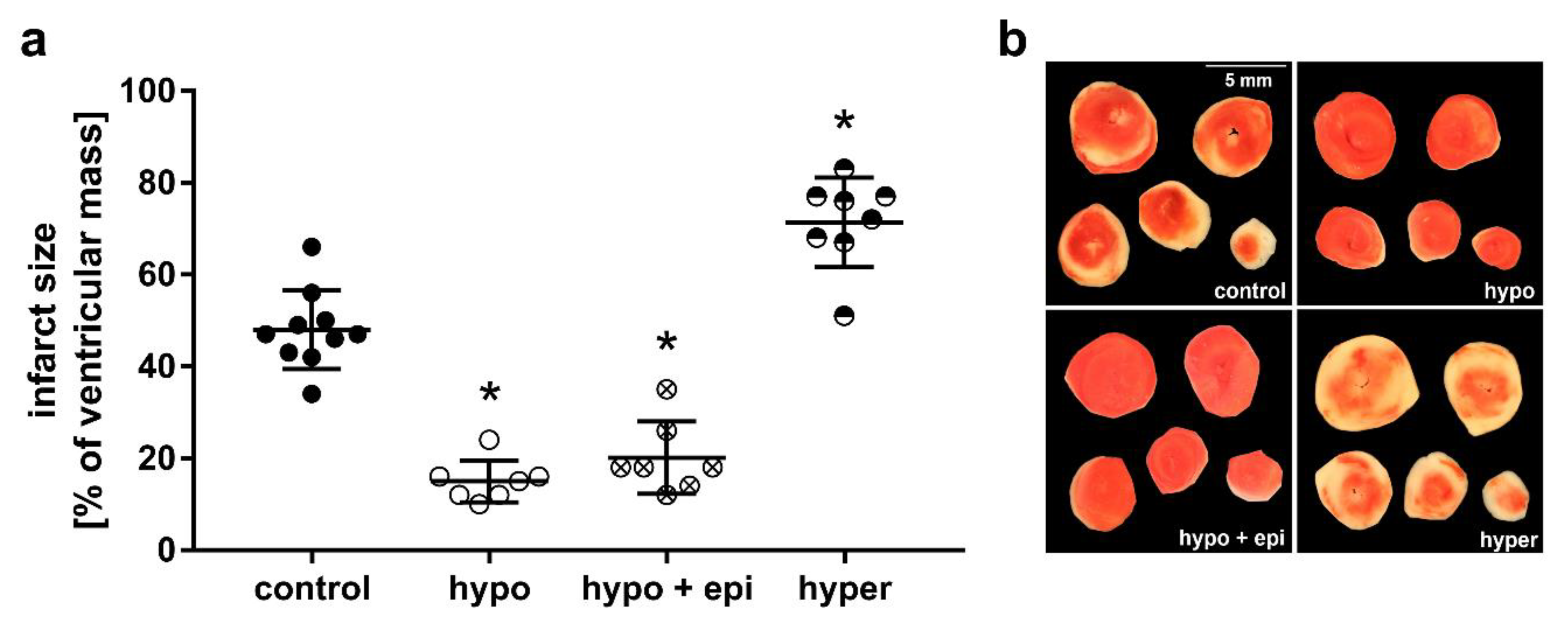
2.2. Impact of Thyroid Dysfunction on Infarct Size
2.3. Impact of Thyroid Dysfunction on Hemodynamics
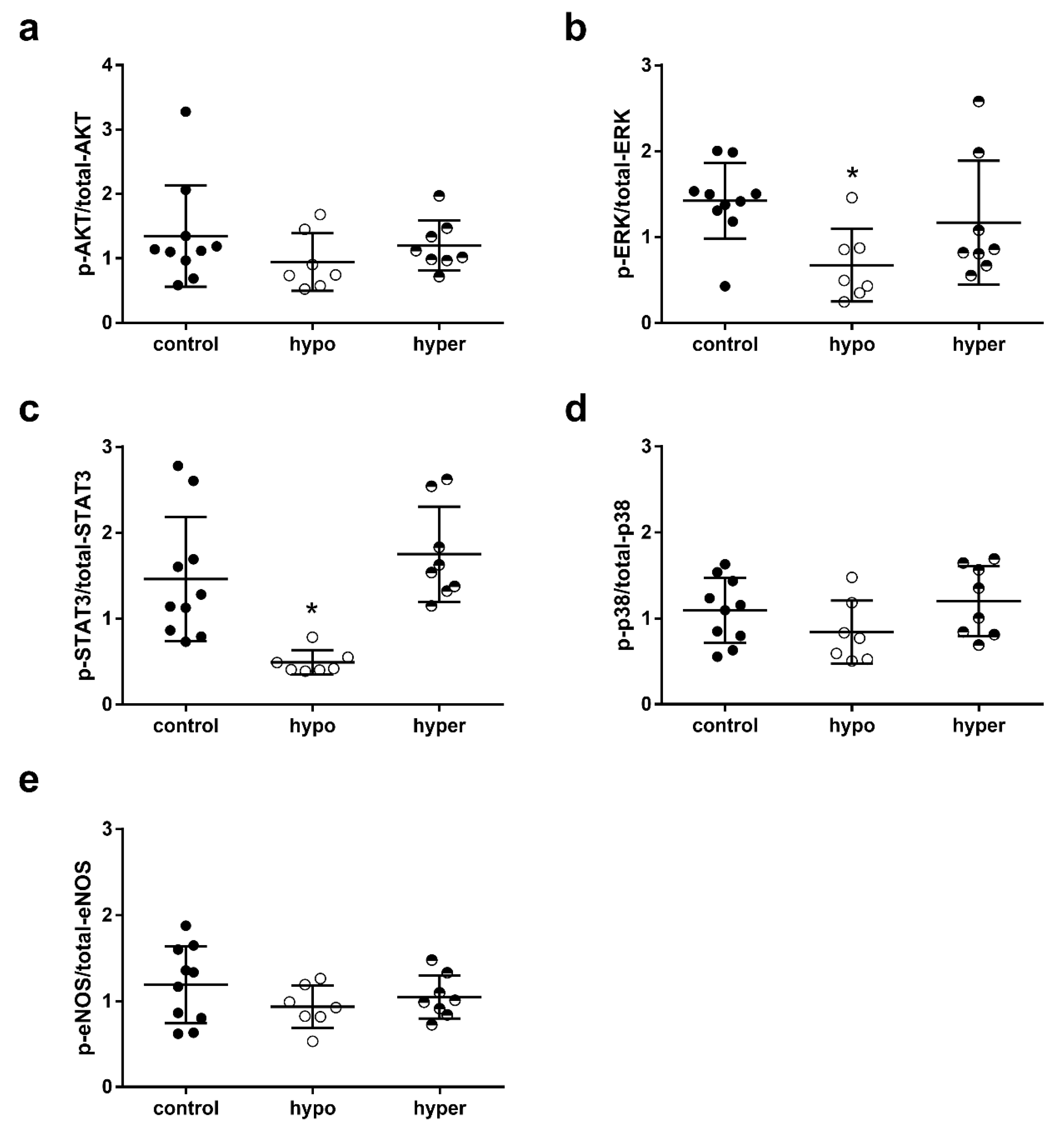
2.4. Impact of Thyroid Dysfunction on Cardioprotective Signaling Pathways
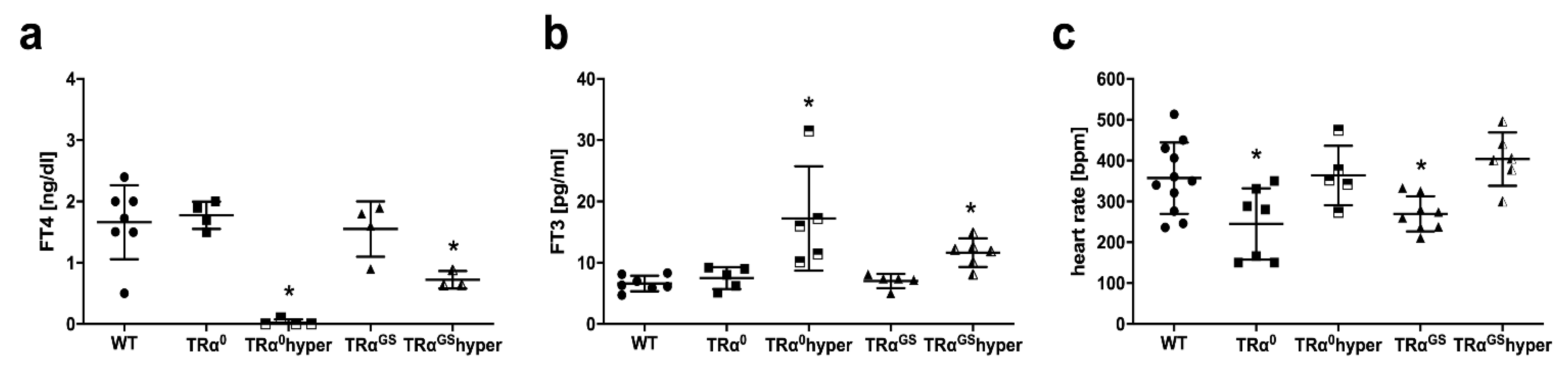
2.5. Serum TH Status and Heart Rate in Mice with Altered TRα Signaling
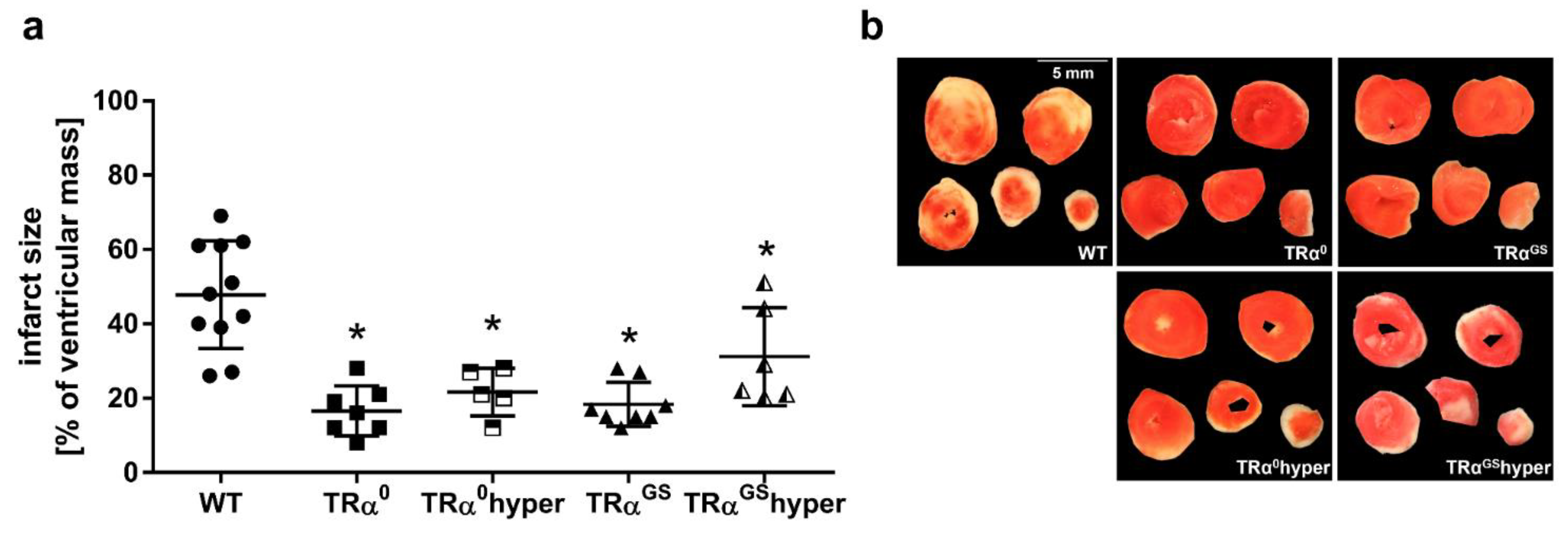
2.6. Impact of TRα Signaling on Infarct Size
2.7. Impact of TRα Signaling on Hemodynamics
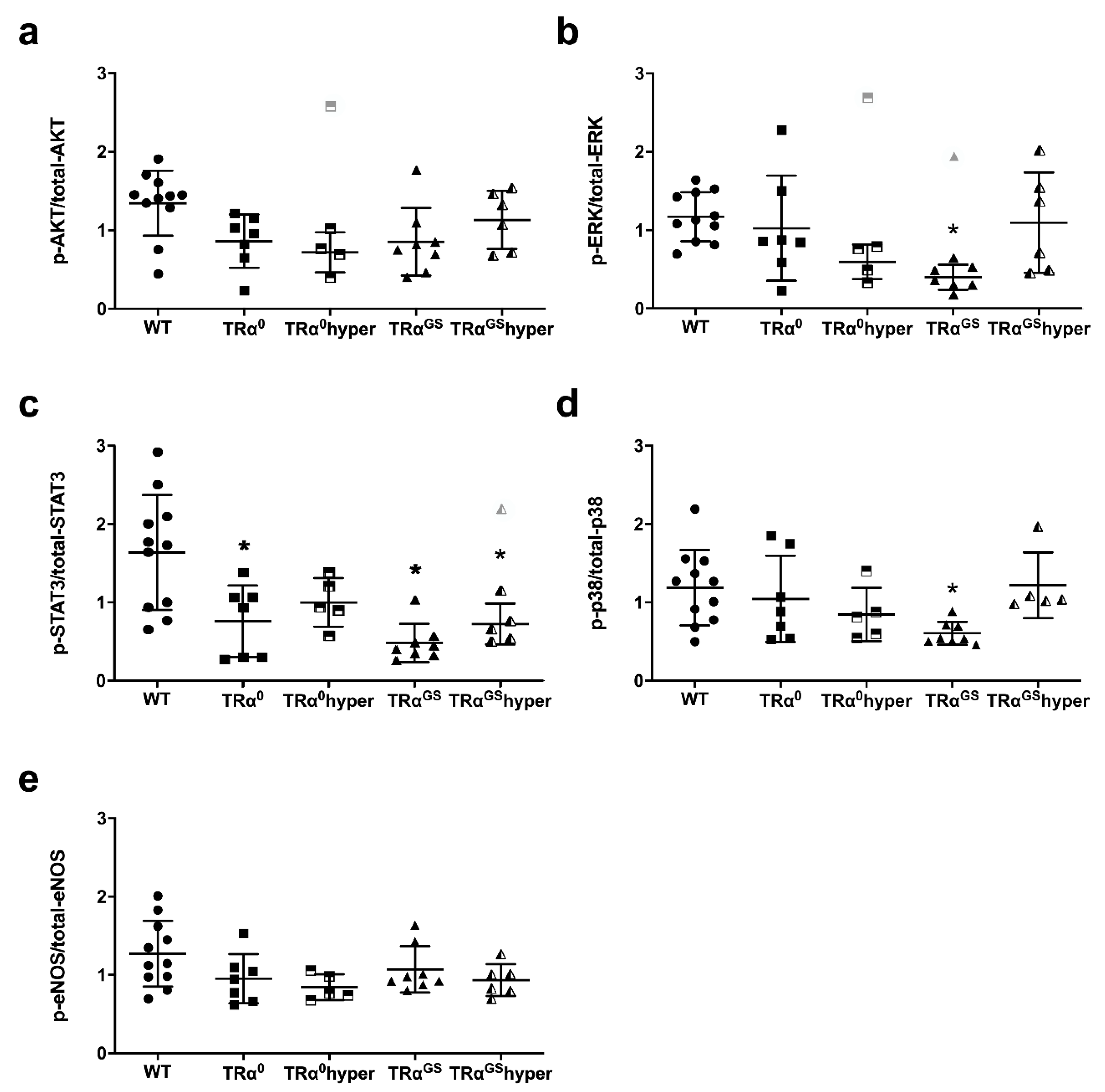
2.8. Impact of Mode of TRα Signaling on Phosphorylation of Key Proteins of RISK, SAFE and eNOS Pathway in Mouse Hearts after Ischemia–Reperfusion
3. Discussion
3.1. Hypothyroidism and Lack of Canonical TRα Signaling Confer Cardioprotection in the Absence of Favorable Hemodynamics and without Canonical Protective Signaling
3.2. Noncanonical TRα Signaling Contributes to Baseline left Ventricular Pressure
3.3. Lack of Canonical TRα Signaling Determines Bradycardia
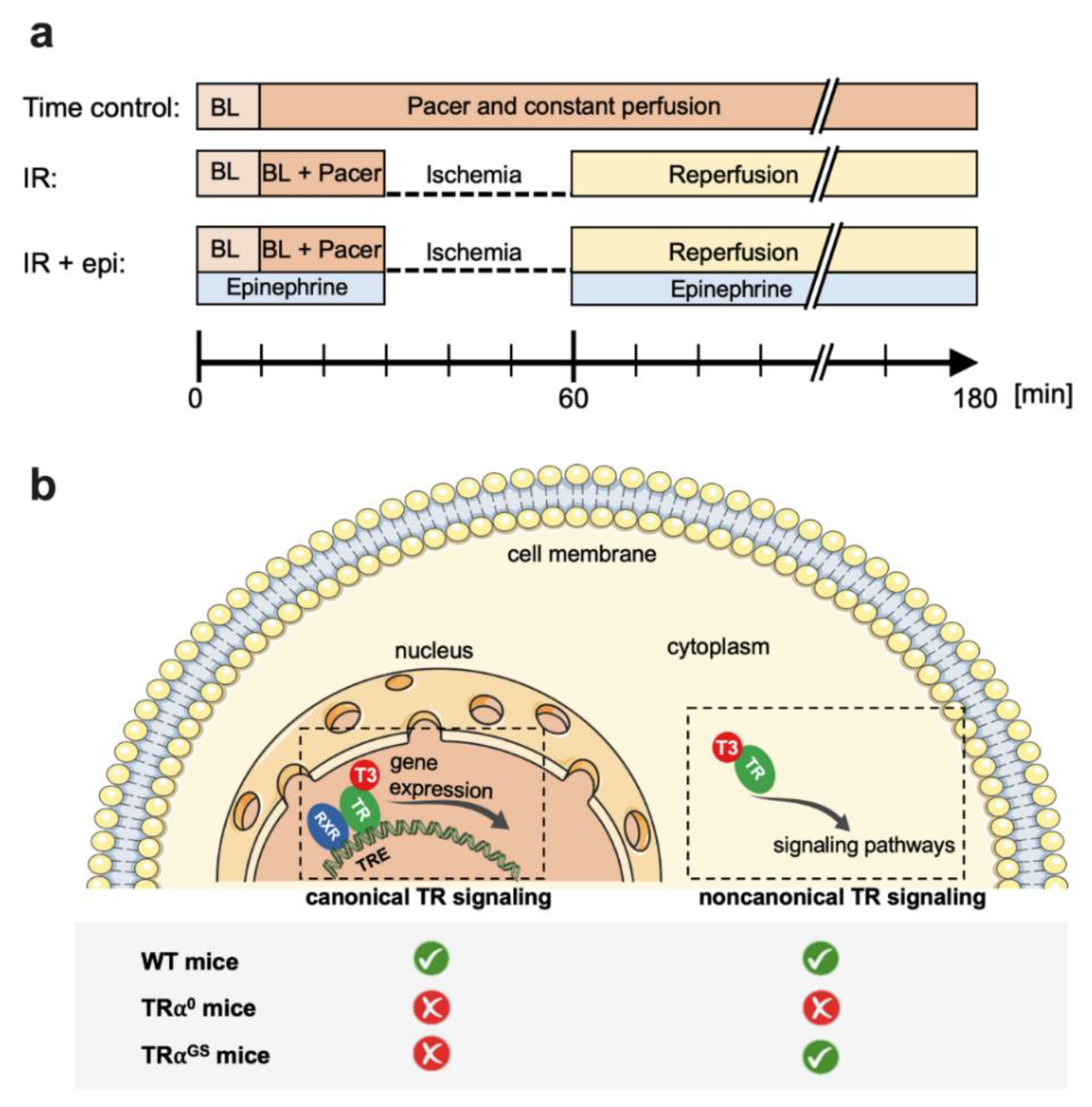
4. Materials and Methods
4.1. Mice and Treatment
4.2. Isolated Mouse Hearts
4.3. Protocols for Isolated Mouse Hearts (Figure 1)
4.4. Infarct Size Determination
4.5. Serum TH Status
4.6. Immunoblot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jabbar, A.; Pingitore, A.; Pearce, S.H.; Zaman, A.; Iervasi, G.; Razvi, S. Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 2017, 14, 39–55. [Google Scholar] [CrossRef]
- Razvi, S.; Jabbar, A.; Pingitore, A.; Danzi, S.; Biondi, B.; Klein, I.; Peeters, R.; Zaman, A.; Iervasi, G. Thyroid Hormones and Cardiovascular Function and Diseases. J. Am. Coll. Cardiol. 2018, 71, 1781–1796. [Google Scholar] [CrossRef]
- Seara, F.A.C.; Maciel, L.; Barbosa, R.A.Q.; Rodrigues, N.C.; Silveira, A.L.B.; Marassi, M.P.; Carvalho, A.B.; Nascimento, J.H.M.; Olivares, E.L. Cardiac ischemia/reperfusion injury is inversely affected by thyroid hormones excess or deficiency in male Wistar rats. PLoS ONE 2018, 13, e0190355. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef] [Green Version]
- Pantos, C.I.; Malliopoulou, V.A.; Mourouzis, I.S.; Karamanoli, E.P.; Paizis, I.A.; Steimberg, N.; Varonos, D.D.; Cokkinos, D.V. Long-term thyroxine administration protects the heart in a pattern similar to ischemic preconditioning. Thyroid 2002, 12, 325–329. [Google Scholar] [CrossRef]
- Venditti, P.; Agnisola, C.; Di Meo, S. Effect of ischemia-reperfusion on heart mitochondria from hyperthyroid rats. Cardiovasc. Res. 2002, 56, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Botker, H.E.; Hausenloy, D.; Andreadou, I.; Antonucci, S.; Boengler, K.; Davidson, S.M.; Deshwal, S.; Devaux, Y.; Di Lisa, F.; Di Sante, M.; et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 39. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L.; Bolli, R.; Canty, J.M., Jr.; Du, X.J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial stunning and hibernation revisited. Nat. Rev. Cardiol. 2021, 18, 522–536. [Google Scholar] [CrossRef]
- Dillmann, W.H. Biochemical basis of thyroid hormone action in the heart. Am. J. Med. 1990, 88, 626–630. [Google Scholar] [CrossRef]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef] [Green Version]
- Flamant, F.; Gauthier, K. Thyroid hormone receptors: The challenge of elucidating isotype-specific functions and cell-specific response. Biochim. Biophys. Acta 2013, 1830, 3900–3907. [Google Scholar] [CrossRef]
- Mourouzis, I.; Kostakou, E.; Galanopoulos, G.; Mantzouratou, P.; Pantos, C. Inhibition of thyroid hormone receptor alpha1 impairs post-ischemic cardiac performance after myocardial infarction in mice. Mol. Cell. Biochem. 2013, 379, 97–105. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I. Thyroid hormone receptor alpha1 as a novel therapeutic target for tissue repair. Ann. Transl. Med. 2018, 6, 254. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I. Translating thyroid hormone effects into clinical practice: The relevance of thyroid hormone receptor alpha1 in cardiac repair. Heart Fail. Rev. 2015, 20, 273–282. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I.; Saranteas, T.; Brozou, V.; Galanopoulos, G.; Kostopanagiotou, G.; Cokkinos, D.V. Acute T3 treatment protects the heart against ischemia-reperfusion injury via TRalpha1 receptor. Mol. Cell. Biochem. 2011, 353, 235–241. [Google Scholar] [CrossRef]
- Flamant, F.; Cheng, S.Y.; Hollenberg, A.N.; Moeller, L.C.; Samarut, J.; Wondisford, F.E.; Yen, P.M.; Refetoff, S. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology 2017, 158, 2052–2057. [Google Scholar] [CrossRef]
- Storey, N.M.; Gentile, S.; Ullah, H.; Russo, A.; Muessel, M.; Erxleben, C.; Armstrong, D.L. Rapid signaling at the plasma membrane by a nuclear receptor for thyroid hormone. Proc. Natl. Acad. Sci. USA 2006, 103, 5197–5201. [Google Scholar] [CrossRef] [Green Version]
- Hones, G.S.; Rakov, H.; Logan, J.; Liao, X.H.; Werbenko, E.; Pollard, A.S.; Praestholm, S.M.; Siersbaek, M.S.; Rijntjes, E.; Gassen, J.; et al. Noncanonical thyroid hormone signaling mediates cardiometabolic effects in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E11323–E11332. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.P.; Marron Fernandez de Velasco, E.; Mizuno, F.; Scappini, E.L.; Gloss, B.; Erxleben, C.; Williams, J.G.; Stapleton, H.M.; Gentile, S.; Armstrong, D.L. A rapid cytoplasmic mechanism for PI3 kinase regulation by the nuclear thyroid hormone receptor, TRbeta, and genetic evidence for its role in the maturation of mouse hippocampal synapses in vivo. Endocrinology 2014, 155, 3713–3724. [Google Scholar] [CrossRef]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantos, C.; Malliopoulou, V.; Mourouzis, I.; Sfakianoudis, K.; Tzeis, S.; Doumba, P.; Xinaris, C.; Cokkinos, A.D.; Carageorgiou, H.; Varonos, D.D.; et al. Propylthiouracil-induced hypothyroidism is associated with increased tolerance of the isolated rat heart to ischaemia-reperfusion. J. Endocrinol. 2003, 178, 427–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapoy-Villanueva, H.; Silva-Platas, C.; Gutierrez-Rodriguez, A.K.; Garcia, N.; Acuna-Morin, E.; Elizondo-Montemayor, L.; Oropeza-Almazan, Y.; Aguilar-Saenz, A.; Garcia-Rivas, G. Changes in the Stoichiometry of Uniplex Decrease Mitochondrial Calcium Overload and Contribute to Tolerance of Cardiac Ischemia/Reperfusion Injury in Hypothyroidism. Thyroid 2019, 29, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, I.B.; Gomes, D.A.; Alenina, N.; Bader, M.; Dos Santos, R.A.; Barreto-Chaves, M.L.M. Cardioprotective effect of thyroid hormone is mediated by AT2 receptor and involves nitric oxide production via Akt activation in mice. Heart Vessels 2018, 33, 671–681. [Google Scholar] [CrossRef]
- Pantos, C.; Malliopoulou, V.; Mourouzis, I.; Thempeyioti, A.; Paizis, I.; Dimopoulos, A.; Saranteas, T.; Xinaris, C.; Cokkinos, D.V. Hyperthyroid hearts display a phenotype of cardioprotection against ischemic stress: A possible involvement of heat shock protein 70. Horm. Metab. Res. 2006, 38, 308–313. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Selmer, C.; Olesen, J.B.; Hansen, M.L.; von Kappelgaard, L.M.; Madsen, J.C.; Hansen, P.R.; Pedersen, O.D.; Faber, J.; Torp-Pedersen, C.; Gislason, G.H. Subclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events: A large population study. J. Clin. Endocrinol. Metab. 2014, 99, 2372–2382. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Skyschally, A.; Gent, S.; Pesch, M.; Heusch, G. STAT3 as a common signal of ischemic conditioning: A lesson on “rigor and reproducibility” in preclinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 3. [Google Scholar] [CrossRef]
- Lieder, H.R.; Kleinbongard, P.; Skyschally, A.; Hagelschuer, H.; Chilian, W.M.; Heusch, G. Vago-Splenic Axis in Signal Transduction of Remote Ischemic Preconditioning in Pigs and Rats. Circ. Res. 2018, 123, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Shih, A.; Davis, F.B.; Davis, P.J. Thyroid hormone promotes the phosphorylation of STAT3 and potentiates the action of epidermal growth factor in cultured cells. Biochem. J. 1999, 338 Pt 2, 427–432. [Google Scholar] [CrossRef]
- Liu, K.L.; Canaple, L.; Del Carmine, P.; Gauthier, K.; Beylot, M.; Lo, M. Thyroid hormone receptor-alpha deletion decreases heart function and exercise performance in apolipoprotein E-deficient mice. Physiol. Genom. 2016, 48, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloss, B.; Trost, S.; Bluhm, W.; Swanson, E.; Clark, R.; Winkfein, R.; Janzen, K.; Giles, W.; Chassande, O.; Samarut, J.; et al. Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor alpha or beta. Endocrinology 2001, 142, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Pazos-Moura, C.; Abel, E.D.; Boers, M.E.; Moura, E.; Hampton, T.G.; Wang, J.; Morgan, J.P.; Wondisford, F.E. Cardiac dysfunction caused by myocardium-specific expression of a mutant thyroid hormone receptor. Circ. Res. 2000, 86, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Macchia, P.E.; Takeuchi, Y.; Kawai, T.; Cua, K.; Gauthier, K.; Chassande, O.; Seo, H.; Hayashi, Y.; Samarut, J.; Murata, Y.; et al. Increased sensitivity to thyroid hormone in mice with complete deficiency of thyroid hormone receptor alpha. Proc. Natl. Acad. Sci. USA 2001, 98, 349–354. [Google Scholar] [CrossRef]
- Wikstrom, L.; Johansson, C.; Salto, C.; Barlow, C.; Campos Barros, A.; Baas, F.; Forrest, D.; Thoren, P.; Vennstrom, B. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. EMBO J. 1998, 17, 455–461. [Google Scholar] [CrossRef]
- Engels, K.; Rakov, H.; Zwanziger, D.; Hones, G.S.; Rehders, M.; Brix, K.; Kohrle, J.; Moller, L.C.; Fuhrer, D. Efficacy of protocols for induction of chronic hyperthyroidism in male and female mice. Endocrine 2016, 54, 47–54. [Google Scholar] [CrossRef]
- Rakov, H.; Engels, K.; Hones, G.S.; Strucksberg, K.H.; Moeller, L.C.; Kohrle, J.; Zwanziger, D.; Fuhrer, D. Sex-specific phenotypes of hyperthyroidism and hypothyroidism in mice. Biol. Sex Differ. 2016, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, H.A.; Kreienkamp, V.; Gent, S.; Kahlert, P.; Heusch, G.; Kleinbongard, P. Kinetics and Signal Activation Properties of Circulating Factor(s) From Healthy Volunteers Undergoing Remote Ischemic Pre-Conditioning. JACC Basic Transl. Sci. 2016, 1, 3–13. [Google Scholar] [CrossRef]
- Skyschally, A.; Kleinbongard, P.; Lieder, H.; Gedik, N.; Stoian, L.; Amanakis, G.; Elbers, E.; Heusch, G. Humoral transfer and intramyocardial signal transduction of protection by remote ischemic perconditioning in pigs, rats, and mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H159–H172. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Size | Baseline | Pacer Baseline | isch5 | isch25 | rep10 | rep20 | rep30 | rep40 | rep50 | rep60 | ||
| CF [mL/min] | control | 10 | 2.4 ± 0.9 | 2.7 ± 0.9 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.9 ± 1.0 | 2.9 ± 1.3 | 2.9 ± 1.3 | 2.8 ± 1.4 | 2.8 ± 1.4 | 2.7 ± 1.3 |
| hypo | 7 | 1.3 ± 0.3 * | 1.1 ± 0.3 * | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.0 ± 0.8 | 1.5 ± 0.6 | 1.4 ± 0.6 | 1.5 ± 0.5 | 1.4 ± 0.5 | 1.4 ± 0.5 | |
| hypo + epi | 7 | § 2.2 ± 0.6 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.9 ± 1.0 | 3.0 ± 0.8 | 3.0 ± 0.8 | 3.0 ± 0.8 | 3.0 ± 0.8 | 2.9 ± 0.8 | ||
| hyper | 8 | 3.0 ± 0.9 | 2.9 ± 0.8 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.4 ± 0.6 | 2.3 ± 0.6 | 2.3 ± 0.6 | 2.3 ± 0.7 | 2.2 ± 0.7 | 2.2 ± 0.6 | |
| WT | 11 | 2.4 ± 0.9 | 2.7 ± 0.9 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.9 ± 1.0 | 2.9 ± 1.3 | 2.9 ± 1.3 | 2.8 ± 1.4 | 2.8 ± 1.4 | 2.7 ± 1.3 | |
| TRα0 | 7 | 1.6 ± 0.9 | 1.6 ± 1.0 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 1.5 ± 0.9 | 1.5 ± 0.9 | 1.5 ± 0.9 | 1.6 ± 0.9 | 1.6 ± 0.8 | 1.7 ± 0.9 | |
| TRα0hyper | 5 | 2.4 ± 0.4 | 2.6 ± 0.3 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.9 ± 0.6 | 2.8 ± 0.5 | 2.8 ± 0.4 | 2.7 ± 0.4 | 2.5 ± 0.4 | 2.4 ± 0.4 | |
| TRαGS | 8 | 2.5 ± 1.5 | 2.6 ± 1.2 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.9 ± 1.4 | 2.8 ± 1.4 | 2.7 ± 1.4 | 2.7 ± 1.4 | 2.6 ± 1.4 | 2.6 ± 1.3 | |
| TRαGShyper | 6 | 1.9 ± 0.5 | 2.0 ± 0.5 | 0.0 ± 0.0 # | 0.0 ± 0.0 # | 2.2 ± 0.6 | 2.1 ± 0.6 | 2.1 ± 0.6 | 2.1 ± 0.7 | 2.1 ± 0.7 | 2.0 ± 0.6 | |
| Group Size | Baseline | Pacer Baseline | isch5 | isch25 | rep10 | rep20 | rep30 | rep40 | rep50 | rep60 | ||
| LVDP [mmHg] | control | 10 | 88 ± 14 | 91 ± 19 | 0 ± 0 # | 0 ± 0 # | 11 ± 19 # | 35 ± 27 # | 42 ± 28 # | 42 ± 27 # | 43 ± 26 # | 43 ± 24 # |
| hypo | 7 | 48 ± 12 * | 30 ± 8 * | 0 ± 0 # | 0 ± 0 # | 20 ± 14 | 23 ± 12 | 27 ± 15 | 26 ± 15 | 29 ± 16 | 29 ± 17 | |
| hypo + epi | 7 | § 97 ± 15 | 0 ± 0 # | 0 ± 0 # | 63 ± 24 * | 69 ± 22 | 76 ± 21 | 77 ± 21 | 75 ± 19 | 73 ± 17 | ||
| hyper | 8 | 122 ± 22 * | 110 ± 20 | 0 ± 0 # | 0 ± 0 # | 2 ± 2 # | 9 ± 22 # | 16 ± 31 # | 22 ± 33 # | 25 ± 35 # | 25 ± 34 # | |
| WT | 11 | 88 ± 14 | 91 ± 19 | 0 ± 0 # | 0 ± 0 # | 11 ± 19 # | 35 ± 27 # | 42 ± 28 # | 42 ± 27 # | 43 ± 26 # | 43 ± 24 # | |
| TRα0 | 7 | 104 ± 25 | 55 ± 23 * | 0 ± 0 # | 0 ± 0 # | 40 ± 27 * | 47 ± 21 * | 49 ± 19 | 50 ± 17 | 52 ± 16 | 51 ± 15 | |
| TRα0hyper | 5 | 104 ± 11 | 96 ± 9 | 0 ± 0 # | 0 ± 0 # | 18 ± 20 # | 53 ± 23 *# | 61 ± 21 *# | 63 ± 14 *# | 64 ± 10 *# | 64 ± 7 *# | |
| TRαGS | 8 | 106 ± 19 | 90 ± 21 | 0 ± 0 # | 0 ± 0 # | 73 ± 25 * | 82 ± 19 * | 81 ± 19 * | 80 ± 15 * | 79 ± 14 * | 76 ± 14 * | |
| TRαGShyper | 6 | 84 ± 24 | 81 ± 20 | 0 ± 0 # | 0 ± 0 # | 16 ± 14 # | 31 ± 28 # | 40 ± 28 # | 43 ± 28 # | 45 ± 28 # | 46 ± 27 # | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pape, J.; Kerp, H.; Lieder, H.R.; Geist, D.; Hönes, G.S.; Moeller, L.C.; Kleinbongard, P.; Führer, D. Cardioprotection by Hypothyroidism Is Not Mediated by Favorable Hemodynamics—Role of Canonical Thyroid Hormone Receptor Alpha Signaling. Int. J. Mol. Sci. 2022, 23, 13340. https://doi.org/10.3390/ijms232113340
Pape J, Kerp H, Lieder HR, Geist D, Hönes GS, Moeller LC, Kleinbongard P, Führer D. Cardioprotection by Hypothyroidism Is Not Mediated by Favorable Hemodynamics—Role of Canonical Thyroid Hormone Receptor Alpha Signaling. International Journal of Molecular Sciences. 2022; 23(21):13340. https://doi.org/10.3390/ijms232113340
Chicago/Turabian StylePape, Janina, Helena Kerp, Helmut R. Lieder, Daniela Geist, Georg Sebastian Hönes, Lars C. Moeller, Petra Kleinbongard, and Dagmar Führer. 2022. "Cardioprotection by Hypothyroidism Is Not Mediated by Favorable Hemodynamics—Role of Canonical Thyroid Hormone Receptor Alpha Signaling" International Journal of Molecular Sciences 23, no. 21: 13340. https://doi.org/10.3390/ijms232113340
APA StylePape, J., Kerp, H., Lieder, H. R., Geist, D., Hönes, G. S., Moeller, L. C., Kleinbongard, P., & Führer, D. (2022). Cardioprotection by Hypothyroidism Is Not Mediated by Favorable Hemodynamics—Role of Canonical Thyroid Hormone Receptor Alpha Signaling. International Journal of Molecular Sciences, 23(21), 13340. https://doi.org/10.3390/ijms232113340